Day 2 :
Keynote Forum
Timothy A. Cross
Florida State Univ. & National High Magnetic Field Lab, USA
Keynote: Membrane Protein Structure, Dynamics & Function: Oriented Sample and Magic Angle Sample Spinning Solid State NMR

Biography:
Timothy Cross has more that 30 years of experience characterizing membrane proteins in lipid bilayer environments using solid state NMR of liquid crystalline bilayer preparations of peptides and proteins. This has brought to light a fundamental understanding of membrane protein biophysics that has led to detailed functional characterizations of membrane channels – in particular the gramicidin monovalent cation channel and the influenza A M2 proton channel. In both systems the unique features of the membrane environment play crucial roles in the functional mechanisms and kinetics of ion conductance. Is this understanding of the influence of membrane and lipid environments through the use of solid state NMR that has driven his research at the frontier of membrane protein biophysics.
Abstract:
Statement of the Problem: Unlike water soluble proteins that have a relatively homogeneous environment, membrane proteins exist in a dramatically heterogeneous environment. The result is protein structure that is stabilized by a different balance of molecular interactions for the membrane embedded portion of the protein compared to the water soluble or membrane interfacial regions of the protein. The result is a need to model the membrane environment as closely as possible to that of the native environment for structural, dynamic and functional characterizations. Methodology: Biological solid state NMR provides a unique opportunity to model the membrane environment with liquid crystalline lipid bilayers and a wide variety of lipids. The samples can be prepared either as liposomes for magic angle sample spinning (MAS) or as uniformly oriented samples (OS) for the spectroscopy. The former provides solution like spectra for both distance and isotropic chemical shift restraints, while oriented samples provide absolute restraints that restrain the atomic sites in the protein structure to the bilayer normal. In addition to structural restraints it is possible to characterize the protein’s dynamics and kinetic rates. Findings, Conclusion & Significance: The structure, dynamics and kinetics associated with the M2 proton channel from influenza A have been characterized yielding a unique mechanism for proton transport by this important drug target. In addition, the cholesterol binding to M2 has been found to stabilize the amphipathic helix in the lipid interface an essential feature for this protein’s functional role in viral budding. Recent structural studies of the CrgA protein from Mycobacterium tuberculosis have characterized a dimeric structure stabilized primarily by intermolecular b-sheet in the membrane interfacial region. The protein is part of the cell division apparatus and appears to play a role in recruiting multiple proteins to the divisome, potentially through its transmembrane domain.
References:
1. Zhou, H.-X. & Cross, T.A. (2013) “Influences of Membrane Mimetic Environments on Membrane Protein Structures” Annual Reviews of Biophysics 42:361-392.
2. Sharma, M., Yi, M., Dong, H., Qin, H., Petersen, E., Busath, D.D., Zhou, H.-X. & Cross, T.A. (2010) “Insight into the Mechanism of the Influenza A Proton Channel from a Structure in a Lipid Bilayer” Science 330:509-512.
3. Murray, D., Das, N., Cross, T.A. (2013) “Solid State NMR Strategy for Characterizing Native Membrane Protein Structures” Accounts of Chemical Research 46:2172-2181.
4. Miao, Y., Fu, R., Zhou, H.-X. & Cross, T.A. (2015) Dynamic, Short Hydrogen Bonds in Histidine Tetrad of Full-Length M2 H+ Channel Reveals Tetrameric Structural Heterogeneity and Functional Mechanism” Structure 23:2300-2308.
5. Das, N., Dai, J., Hung, I., Rajagopalan, M., Zhou, H.-X. & Cross, T.A. (2015) “Structure of CrgA, a Cell Division Structural and Regulatory Protein from Mycobacterium tuberculosis in Lipid Bilayers” Proc. Natl. Acad. Sci. 112:E119-E126.
Keynote Forum
Wladek Minor
University of Virginia, USA
Keynote: Reproducibility in Biomedical Sciences - Big Data Perspective

Biography:
Wladek Minor is a Harrison Distinguished Professor of Molecular Physiology and Biological Physics at University of Virginia. He is an expert in structural biology and data mining. He is an author of over 190 papers that attracted over 37,000 of citations. His Relative Citations Ratio is above 560.He trained over 80 scientists that currently pursue career in academia, government and industry.
Abstract:
Experimental reproducibility is the cornerstone of scientific research, upon which all progress rests. The veracity of scientific publications is crucial because subsequent lines of investigation rely on previous knowledge. Several recent systematic surveys of academic results published in biomedical journals reveal that a large fraction of representative sets of studies in a variety of fields cannot be reproduced in another laboratory. Big Data approach and especially NIH Big Data to Knowledge (BD2K) program is coming to the rescue.
The goal of the presented research is to provide the biomedical community with a strategy to increase the reproducibility of reported results for a wide range of experiments by building a set of “best practices”, culled by extensive data harvesting and curation combined with experimental verification of the parameters crucial for reproducibility. Experimental verification assisted by the automatic/semi-automatic harvesting of data from laboratory equipment into the already developed sophisticated laboratory information management system (LIMS) will be presented. This data in, information out paradigm will be discussed.
References:
- Zheng H, Cooper DR, Porebski PJ, Shabalin IG, Handing KB, Minor W (2017) CheckMyMetal: a macromolecular metal-binding validation tool. Acta Cryst. D 73: 223-233
- Grabowski M, Minor W (2017) Sharing Big Data. IUCrJ 4: 3-4
- Rupp B, Wlodawer A, Minor W, Helliwell JR, Jaskolski M (2016) Correcting the record of structural publications requires joint effort of the community and journal editors. FEBS J. 283: 4452-4457
- Grabowski M, Langner KM, Cymborowski M, Porebski PJ, Sroka P, Zheng H, Cooper DR, Zimmerman MD, Elsliger MA, Burley SK, Minor W (2016) A public database of macromolecular diffraction experiments. Acta Cryst D 72: 1181-1193
- Niedzialkowska E, Gasiorowska O, Handing KB, Majorek KA, Porebski PJ, Shabalin IG, Zasadzinska E, Cymborowski M, Minor W (2016) Protein purification and crystallization artifacts: The tale usually not told. Protein Science 25: 720-33
Keynote Forum
Yuri L. Lyubchenko
University of Nebraska Medical Center, USA
Keynote: Protein-protein interaction and amyloid cascade hypothesis for Alzheimer’s disease

Biography:
Yuri L. Lyubchenko is Professor of Pharmaceutical Sciences at the University of Nebraska Medical Center, Omaha, NE, USA. His research focuses on understanding fundamental mechanisms underlying health and disease, which are key to developing new and more effective diagnostics and medications. This primarily basic research allows him not only identify new drug targets for small molecule drugs, it also leads to development of the nanotools and methods to discover novel approaches for diagnostic, treatment and disease prevention and to more rapidly determine their efficacy at the molecular level.
Abstract:
The amyloid cascade hypothesis is currently considered as the main model for a vast number of neurodegenerative diseases including Alzheimer’s, Parkinson’s, and Huntington’s diseases. Numerous studies have shown that amyloidogenic proteins are capable of spontaneous assembly into aggregates, and eventually form fibrillar structures found in amyloid or amyloidâ€like deposits. However, there is a serious complication with translating current knowledge on amyloid aggregation in vitro to understand the aggregation process in vivo. If the critical concentration for the spontaneous aggregation of Aβ peptide in vitro is in the micromolar range, physiological concentrations of Aβ are in the low nanomolar range making impossible amyloids to assemble. We have discovered a novel on-surface aggregation pathway that allows for spontaneous assembly of amyloid beta peptides at the physiological concentration range. Our combined experimental and computer modeling approaches demonstrate that the on-surface aggregation is a dynamic process, so the assembled aggregate can dissociate from the surface to the bulk solution. As a result, the dissociated oligomers can play roles of seeds for aggregation in the bulk solution, or start a neurotoxic effect such as phosphorylation of the tau protein to initiate its misfolding and aggregation. Both processes lead to neurodegeneration. Importantly, in the vast majority of cases, we found that aggregates formed on the surface are oligomers, which are considered to be the most neurotoxic amyloid aggregates. Therefore, we posit that on-surface aggregation is the mechanism by which neurotoxic amyloid aggregates are produced under physiological conditions. A change in membrane properties leading to an increase in affinity of amyloid proteins to the membrane surface facilitates the assembly of stable oligomers. The proposed model is a significant departure from the current model as it directs the development of treatments and preventions towards approaches that control the cell membranes properties and composition preventing the on-surface aggregation process.
References:
1. Lyubchenko, Y. L. (2014) Centromere chromatin: a loose grip on the nucleosome?, Nat Struct Mol Biol 21, 8.
2. Lyubchenko, Y. L., and Shlyakhtenko, L. S. (2015) Chromatin imaging with time-lapse atomic force microscopy, Methods Mol Biol 1288, 27-42.
3. Lyubchenko, Y. L., and Shlyakhtenko, L. S. (2015) Atomic Force Microscopy Imaging and Probing of Amyloid Nanoaggregates, In Handbook of Clinical Nanomedicine: From Bench to Bedside (Bawa, R., Audette, G. & Rubinstein, I., Ed.), p 1500. , Pan Stanford Publishing, Singapore. .
4. Sun, Z., Tan, H. Y., Bianco, P. R., and Lyubchenko, Y. L. (2015) Remodeling of RecG Helicase at the DNA Replication Fork by SSB Protein, Sci Rep 5, 9625.
5. Proctor, E. A., Fee, L., Tao, Y., Redler, R. L., Fay, J. M., Zhang, Y., Lv, Z., Mercer, I. P., Deshmukh, M., Lyubchenko, Y. L., and Dokholyan, N. V. (2016) Nonnative SOD1 trimer is toxic to motor neurons in a model of amyotrophic lateral sclerosis, Proc Natl Acad Sci U S A 113, 614-619.
- Track 8: Structural Biology in Complexity Arenas
Session Introduction
Charles W. Carter, Jr
University of North Carolina, USA
Title: How Does Domain Motion Contribute to Transition-State Stabilization? Combinatorial Thermodynamic Cycle Analysis of Conformational Coupling During Tryptophan Activation
Time :

Biography:
Charlie Carter is an X-ray crystallographer who studies the origin, evolution, and structural biology of aminoacyl-tRNA synthetases. His research group introduced the use of Urzymes—highly conserved structural cores that retain large fractions of the transition-state stabilization free enregies of full length enzymes—as experimental models of ancestral enzymes.
Abstract:
Enzyme mechanisms, especially those that couple NTP hydrolysis to mechanical work and information, use sophisticated dynamic networks to transduce active-site chemistry into domain motions that change binding affinities [1]. We measured and cross-validated the energetics of such networks in B. stearothermophilus Tryptophanyl-tRNA synthetase (TrpRS) using both multi-mutant and modular thermodynamic cycles [2]. Coordinated domain motions develop shear in a core packing motif conserved in >125 different protein superfamilies [2]. Multi-dimensional combinatorial mutagenesis showed that four side chains from this “molecular switch” move coordinately with the active-site Mg2+ ion in the transition state for amino acid activation [3]. A modular thermodynamic cycle consisting of full-length TrpRS, the Urzyme, and the Urzyme plus each of the two domains deleted in the Urzyme [4]gives similar energetics. These complementary experiments establish that catalysis and specificity in full-length TrpRS are both coupled by 5 kcal/mole to: (i) the core packing region where domain movement generates shear, and (ii) the simultaneous motion of the two domains relative to the Urzyme. Theory shows that the minimum action path algorithm estimates thermodynamically meaningful contributions of domain movement to kinetic rates [5]. Correlations between those parameters, the experimental rates, and structural variations induced in the combinatorial mutants confirm that these estimates are realistic. These results validate our previous conclusion that catalysis by Mg2+ ion is coupled to the overall domain motion (3). Computational free energy surfaces demonstrate that TrpRS catalytic domain motion itself is endergonic but is driven thermodynamically by PPi release [5,6]. Comparison of the impact of combinatorial mutagenesis on pre-steady state and steady-state rates confirm that dynamic active-site pre-organization endows TrpRS with the elusive conditionality of NTP utilization on domain motion.
References:
- Carter CW, Jr. (2017) High-Dimensional Mutant and Modular Thermodynamic Cycles, Molecular Switching, and Free Energy Transduction. Annual Review of Biophysics 46:In Press. doi:10.1146/annurev-biophys-070816-033811
- Carter CW, Jr., Chandrasekaran SN, Weinreb V, Li L, Williams T (2017) s Structural Dynamics 4:032101
- Weinreb V, Li L, Carter CW, Jr. (2012). Structure, vol 20.
- Li L, Carter CW, Jr (2013) J Biol Chem 288 (29 November):34736–34745. doi:10.1074/jbc.M113.510958
- Chandrasekaran SN, Carter CWJ (2017) Structural Dynamics 4:In press
Christophe Guyeux
Université de Bourgogne Franche-Comté, France
Title: On two ways to predict the protein folding process over a chaotic model
Time :

Biography:
Guyeux has a record of about 120 scholarly publications. Since 2010, he published 43 articles in peer-reviewed international journals (as a co-author, including the top-ranked ones in the areas of computer science and interdisciplinary applications, such as AIP Chaos, Plos One, or Clinical Infectious Diseases). He is co-author of 2 book chapters and 2 scientific monograms. He is also author of 4 software patents, 53 articles that appeared in proceeding of peer-reviewed international conferences. His topics of research encompasses bioinformatics, discrete dynamical systems, and information security.
Abstract:
In our first theoretical studies about folded self-avoiding walks, we have raised raises several questions regarding the protein structure prediction problem and the current ways to solve it. In one category of PSP software, the protein is supposed to be synthesized first as a straight line of amino acids, and then this line of a.a. is folded out until reaching a conformation that optimizes a given scoring function. The second category consider that, as the protein is already in the aqueous solvent, it does not wait the end of the synthesis to take its 3D conformation. So they consider SAWs whose number of steps increases until the number of amino acids of the targeted protein and, at each step, the current walk is stretched (one amino acid is added to the protein) in such a way that the pivot k placed in the position that optimizes the scoring function. We have proven that the two sets of possible conformations are different. So these two kinds of PSP software cannot predict the same kind of conformations.
We have proven too that the folding process G in the 2D model is chaotic according to Devaney. A consequence of this theorem is that this process is highly sensitive to its initial condition. If the 2D model can accurately describe the natural process, then this theorem implies that even a minute difference on an intermediate conformation of the protein, in forces that act in the folding process, or in the position of an atom, can lead to enormous differences in its final conformation. In particular, it seems very difficult to predict, in this 2D model, the structure of a given protein by using the knowledge of the structure of similar proteins. Let us remark that the whole 3D folding process with real torsion angles is obviously more complex. And finally, that chaos refers to our incapacity to make good prediction, it does not mean that the biological process is a random one.
References:
- Christophe Guyeux, Nicod J M, Philippe L, and Bahi J (2015) The study of unfoldable self-avoiding walks: Application to protein structure prediction software. Journal of Bioinformatics and Computational Biology 13(4): 1550009.
- Christophe Guyeux, Côté N-L, Bienia W and Bahi J (2013) Is protein folding problem really a NP-complete one? First investigations. Journal of Bioinformatics and Computational Biology 12(1): 24.
- Christophe Guyeux, Bahi J, Mazouzi K, and Philippe L (2013) Computational investigations of folded self-avoiding walks related to protein folding. Computational Biology and Chemistry 47: 246-256.
- Christophe Guyeux, Bahi J, Côté N and Salomon M (2012) Protein folding in the 2D hydrophobic-hydrophilic (HP) square lattice model is chaotic. Cognitive Computation 4(1): 98-114.

Biography:
Marta obtained Ph.D. in 2007 in nanomaterials from Cranfield University. In 2011, she joined Structural Biology group led by Alaistair Lawson at UCB. For the past five years she has been developing expertise in small molecule drug discovery using a fragment based approach. She had also been involved in the assay development for a novel antibody-enable drug discovery approach. Prior to joining UCB she was awarded a Postdoctoral Fellowship at the Institute of Food Research in Norwich to study the mono and multilayer films used to encapsulate active ingredients for a controlled and site-specific delivery within the GIT. During her time at IFR she worked on numerous projects including a commercial project for Pfizer focused on AFM imaging of proteins and polysaccharides constituting vaccines. She has also been part of the team under the leadership of Claudio Nicolletii investigating the outcome of consumption of the probiotic L. casei Shirota in subjects with Seasonal Allergic Rhinitis.
Abstract:
Protein-protein interactions (PPIs) are of critical importance in the majority of biological cellular processes including DNA repair, immune, and allergic responses. Despite their therapeutic relevance, PPIs are intrinsically challenging targets due to complexity of interactions, assay tractability and the lack of well-defined binding pockets at the interacting surfaces.
In the quest for small molecule drug candidates targeting PPIs, there have been a number of different approaches adopted, which include: use of existing lead or drugs, natural products, high-throughput screening and more recently established powerful fragment-based drug discovery.
At UCB we have integrated the fragment-based methodology with biological and structural information obtained from antibody-validated protein targets, to develop specific small molecule inhibitors of PPIs. An ensemble of biophysical methods (i.e. SPR, ITC, FRET, MS and ligand-based NMR), corroborated by functional data, were employed to identify and validate fragment hits that constituted the starting point for our PPI inhibitor drug discovery programs. We have also employed antibodies as research tools to hold target proteins in biologically active conformations, aiding the discovery of new small molecules for challenging targets. By holding the target protein in biologically relevant conformations, new sites (in particular allosteric sites), which would otherwise be inaccessible, may become available for binding. The ability to capture the target protein in a specific conformation with high affinity for a significantly long time opens the possibility for a small-fragment molecule screening.
Ulf Skoglund
Okinawa Institute of Science and Technology, Japan
Title: Structure of Human IgM in complex with the Malaria protein PfEMP1

Biography:
Ulf Skoglund, born 1950, received his Ph.D. in 1969 at Stockholm University, Sweden. He was a Professor 1996 – 2009 at Karolinska Institutet, Stockholm, Sweden. Since 2010, he is a Professor in Structural Cellular Biology at Okinawa Institute of Science and Technology, Okinawa, Japan.
He has developed electron-tomographic technologies allowing for images of proteins to be generated so that e.g. X-ray structures can be fitted into the 3D densities. This technique is termed COMET (constrained maximum entropy tomography). His Unit has also developed a large-scale dynamics method that allows for quantitative calculations of molecular movements in solution. Current developments concern the mathematics and improvements of the basic 3D reconstruction principles, as well as work on reconfigurable and high performance computing. His Unit has also been actively pursuing several cell biology projects.
Abstract:
Children under the age of 5 years have huge malaria burden in endemic area. Increased death in complicated malaria is due to increased sequestration to tissues and agglutination with erythrocytes and cells of our immune system. It is known that parasites that bind to non-immune IgM cause severe malaria due to increased rosetting (agglutination). Using biochemical, parasitology and electron tomography techniques we have identified that
PfEMP1, a crescent shaped molecule interacts with human IgM through its bulky C-terminus (membrane proximal) in 1:1 and 2:1 ratio. While the bulky C terminus limits the stoichiometry of this interaction yet clusters parasite molecule PfEMP1 (P. falciparum Erythrocyte Membrane Protein-1) to mediate robust host parasite interaction. Structural analysis revealed that PfEMP1 could also preclude the activation of complement mediated lysis of parasite in spite of IgM deposition on parasitized RBC surface. We also found that IgM although not a rosetting factor enhances this interaction by increasing the strength of this interaction by at least four fold. In terms of physiological relevance, we need to understand that new born babies have high level of IgM and could be more prone to agglutination and hence more deaths due to malaria.
Matthias Buck
Case Western Reserve University, USA
Title: Solution NMR relaxation and µs Molecular Dynamics Simulations of Dynamic Protein-Protein and Protein-Membrane Complexes
Time :

Biography:
The Buck laboratory studies two receptor families responsible for cell guidance and positional maintenance (Plexins and Ephrins), both with key involvement in cardiovascular and neuronal development and disease, esp. cancer. We use a wide range of structural biology (solution NMR / x-ray crystallography) and protein biophysical tools (CD, fluorescence spectroscopy, ITC and SPR) in a problem oriented approach. Part of the laboratory also pursues computational modeling and molecular dynamics to provide additional perspective on the problems, provide new insights into the experimental data and to suggest further studies. Small GTPases and their interaction with the plexin receptor cytoplasmic domains has been a major focus of the laboratory and recently we have become very interested in protein-membrane interactions; both the transmembrane regions of the receptors as well as the transient interactions of receptor and GTPase domains with membranes
Abstract:
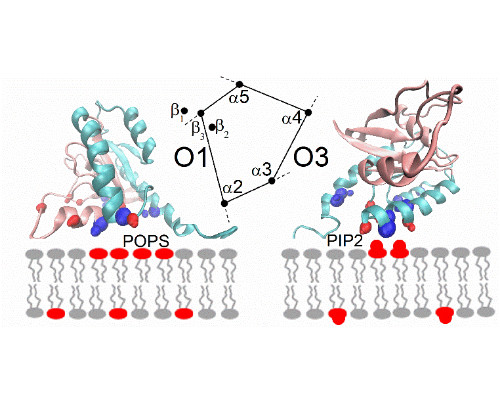
It is now recognized that protein-protein interactions in solution are often dynamic, especially if the binding affinities are only moderately strong. Dynamics originate in part from the interconversion between structures of the protein complex, e.g. one bound state that is in equilibrium with one or several alternate configurations. We determined the structure of such a complex using NMR restraints [1] and saw the transitions between different configurations in microsecond length all-atom molecular dynamics simulations [2]. Recently, we also studied the dissociation process of mutant complexes which had a weakened primary interaction interface. Those simulations suggested that there is no single dissociation pathway, but that the separation first involves transitions to binding interfaces with fewer/weaker contacts [3].
Comparison is made between experimental NMR relaxation measurements on the ps-ns as well as µs-ms timescale with the microsecond all atom simulations, also in the context of new simulations of the protein association process. The functional significance of the protein complex alternate states and their dynamics are discussed.
In a second part of the presentation, we consider a second system involving transient interactions; this time between K-Ras and the lipid bilayer of the plasma membrane [4]. Our recent simulations the full length GTPase at different membranes reveal the underlying rules of the interactions, emphasizing electrostatic contacts but also protein topology [5]. Again, simulations are compared with NMR experiments, carried out at model systems for the membrane.
References:
- Lee, HJ, Hota, P.K, Chugha, P., Miao, H., Zhang, L, Kim, SJ, Alviani, R.S, Stetzig, L., Wang, B. and Buck, M. (2012) “NMR structure of a hetero-dimeric SAM: SAM complex: Characterization and manipulation of EphA2 binding reveal new cellular functions of SHIP2”. Structure 20, 41-55.
- Zhang, L., and Buck, M. (2013) "Molecular Simulations of a Dynamic Protein Complex: Role of Salt-Bridges and Polar Interactions in Configurational Transitions" Biophys. J. 105, 2412-2417
- Zhang, L., Borthakur, S. and Buck, M. (2016) "Dissociation of a Dynamic Protein Complex studied by All Atom Molecular Simulation" Biophys. J., 110, 877-886.
- Li, Z., Cao, S., & Buck, M. (2016) “K-Ras at an Anionic Membrane: Orientation, orientation...orientation. Insights from recent simulation and experimental work”. New&Notable in Biophys. J. 110, 1033-1035.
- Li, Z. & Buck, M. (2017) “Computational Modelling Reveals Signaling Lipids Modulate the Orientation of K-Ras4A at the Membrane Reflecting Protein Topology.” Structure, revision submitted.
Beat Vögeli
University of Colorado at Denver, USA
Title: Functional protein conformation networks probed by NMR nanorulers
Time :

Biography:
Beat Vögeli has his expertise in nuclear magnetic resonance (NMR) spectroscopy of biomacromolecules. He develops methodology for the elucidation of conformation and communication networks within and between proteins and nucleic acids. He received his Ph.D. degree at the ETH Zürich in the group of Konstantin Pervushin. After a postdoctoral stay at the National Institutes of Health, Bethesda USA, in the group of Ad Bax, he returned to ETH Zürich to become Oberassistant in the group of Roland Riek and Privatdozent. He is currently an assistant professor at the University of Colorado at Denver in the Department of Biochemistry and Molecular Genetics.
Abstract:
The function of a protein is tightly connected to its conformational network. Often, subtle differences distinguish interchanging states with distinct properties. One major challenge in structural biology is a sufficiently complete description of the structural landscape and the exchange dynamics between structural states at atomic resolution. We have replaced the standard NMR structure determination by an approach that generates multi-state ensembles from a dense network of tight averaged distance restraints derived from exact measurements of nuclear Overhauser enhancements (eNOEs) [Vögeli 2014, Vögeli et al. 2016].
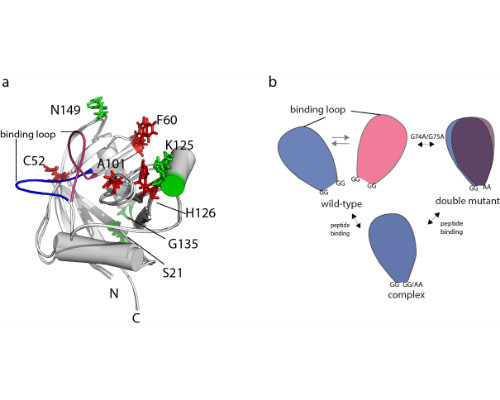
Here, we present the identification of conformational networks harbored by the two human cis/trans isomerases cyclophilin A and Pin1 using the ‘nanorulers’ provided by eNOEs.
We have previously presented an eNOE-based ensemble description of cyclophilin that reveals the presence of a closed and an open state, the latter of which preorganizes the catalytic site for catalysis [Chi et al. 2015]. Based on this finding, we demonstrate here a ligand-selective change of the binding affinity to the active site by tuning of the dynamics of a highly flexible loop [Vögeli et al. 2016]. We show that the binding affinity is increased upon substitution of double glycines to alanines at either of the hinge regions of a loop. The equilibrium distribution is shifted towards more binding-competent conformations.
Comparison of the eNOE-based ensembles of the free and ligand-bound WW domain of Pin1 reveals a conformational network that extends into the interface formed with the enzymatically active PPIase domain. This finding may offer an atomic-picture explanation for the previously discovered communication between the two domains [Peng 2015].
References:
- Vögeli B, Kazemi S, Güntert P, Riek R (2012) Spatial elucidation of motion in proteins by ensemble-based structure calculation using exact NOEs. Nat Struct Mol Biol 19:1053-1057.
- Vögeli B, Olsson S, Güntert P, Riek R (2016) The exact NOE as an alternative in ensemble structure determination. Biophys J 110:113-126.
- Chi C, Vögeli B, Bibow S, Güntert P, Riek R (2015) A structural ensemble for the enzyme cyclophilin reveals an orchestrated mode of action at atomic resolution. Angew Chem Int Ed Engl 54:11657-11661.
- Vögeli B, Bibow S, Chi C (2016) Enzyme selectivity fine-tuned through dynamic control of a loop. Angew Chem Int Ed Engl 55:3096-30100.
- Peng J (2015) Investigating dynamic interdomain allostery in Pin1. Biophys Rev 7:239-249
Shin-ichi Tate
Hiroshima University, JAPAN
Title: Inter-domain communication through intrinsically disordered region (IDR) revealed through the ensemble structure analysis
Time :

Biography:
Abstract:
The functionally relevant inter-domain communication between the domains linked by intrinsically disordered region (IDR) was explored by NMR in combination with small angle X-ray scattering (SAXS). Based on the ensemble structure analyses and the numerical simulations to reproduce the chemical shift changes along with the substrate concentration, we have demonstrated how the domains cooperate to enhance the protein function through the substantially dynamic spatial allocation of the domains.
Pin1, a proline cis/trans isomerase, comprises two domains linked by 10-residue IDR; one is the substrate biding domain to recognize pSer/pThr-Pro motif and the other is the enzyme domain that rotates the Pro peptide bond in the motif. The enzyme domain shows very limited affinity to the substrate, but its binding ability was enhanced by two orders of magnitude in the presence of the substrate binding domain linked by IDR; in which the inter-domain ‘fly-casting’ mechanism plays to keep the substrate bound to Pin1 by tossing and receiving the substrate between the domains, once the substrate in bound to either one of the domains. A new functional aspect of IDR will be addressed.
References:
- Tochio,N., Umehara,K., Uewaki,J., Flechsig,H., Kondo,M., Dewa,T., Sakumar,T., Yamamoto,T., Saitoh,T, Togashi,Y., and Tate,S. (2016): Non-RVD mutations that enhance the dynamics of the TAL repeat array along the superhelical axis improve TALEN genome editing efficacy, Scientific Reports, 6, 37887.
- Wang,J., Tochio,N., Kawasaki,R., Tamari,Y., Xu,N., Uewaki,J., Utsunomiya-Tate, N., and Tate,S. (2015): Allosteric breakage of the hydrogen bond within the dual-histidine motif in the active site of human Pin1 PPIase, Biochemistry, 54, 5242-5253.
- Xu,N. Tochio,N., Wang,J., Tamari,Y., Uewaki,J. Utsunomiya-Tate,N., Igarashi,K., Shiraki,T., Kobayashi,N., and Tate,S. (2014): The C113D mutation in human Pin1 causes allosteric structural changes in the phosphate binding pocket of the PPIase domain through the tug of war in the dualhistidine motif, Biochemistry, 53, 5568-5578.
- Hashimoto,M., Kodera,N., Tsunaka,Y., Oda,M., Tanimoto,M., Ando,T., Morikawa,K., and Tate,S. (2013): Phosphorylation-coupled intramolecular dynamics of unstructured regions in chromatin remodeler FACT, Biophys,J. 104, 2222-2234.
- Mizuno, S., Amida, H., Kobayashi, N., Aizawa, S. and Tate, S. (2011): The NMR structure of FliK, the trigger for the switch of substrate specificity in the flagellar type â…¢ secretion apparatus. J. Mol. Biol., 409, 558-573
Arieh Zaritsky
Ben-Gurion University of the Negev, Israel
Title: Is Nucleoid Complexity hence Cell Diameter Limited by the Eclipse?
Biography:
Arieh Zaritsky of Ben-Gurion University's Faculty of Natural Sciences (http://ariehz.weebly.com/) runs a laboratory investigating parallel fields, pure and applied. During his career at BGU (1973-todate), Dr. Zaritsky has instructed over 50 trainees (graduate students and scientists) and was awarded numerous research grants, allowing him to study both fields of expertise. He visited many higher education Institutions around the world and delivered invited lectures related to both research fields at international meetings. After obtaining a distinguished MSc in Genetics at The Hebrew University of Jerusalem (1967), he graduated at Leicester University (1971) and post-doc'ed at The Copenhagen's Institute of Microbiology (1972). Professor Zaritsky is a recognized expert on bacterial cell physiology and bacteriophage multiplication and published over 130 peer-reviewed articles (http://ariehz.weebly.com/articles.html). Dr. Zaritsky Chaired BGU's Life Sciences department (1989-1991) and is an Editorial Board member of Bioengineered and awardee of 1994 Burroughs-Wellcome/ASM Visiting Professorship.
Abstract:
Cell width W of Escherichia coli is correlated with the mean complexity of its nucleoid, which is expressed as the ratio between the mean times to replicate it and to duplicate the cell aka the number of replication positions n. A set of old, puzzling observations of cell size and dimensions is qualitatively consistent with the view that W is determined by n, and that branching results from breaching a maximum possible value. This maximum nmax is interpreted in terms of a minimal distance possible between successive moving replisomes, so-called eclipse. The data is subject to analytical quantification designed to model the correlations in a way that may (1) shed light on the necessary coupling between the two unique structures in a bacterial cell, nucleoid and sacculus, and (2) lead to decipher the primary signal transduced from DNA to the peptidoglycan biosynthetic pathway. The first approximation is not sufficient to account for the rate at which average cell size rises with time (Po-Yi H and Amir A, personal communication), hence two additional causes are considered to reconcile this discrepancy: loss of division capacity of some DNA-less cells and dependence of the time needed for division on W. A physical signal is invoked, related to transcription/translation of membrane protein genes coupled to membrane-insertion of these proteins termed "transertion", but means to measure the reciprocal stress imposed by transertion strings on both nucleoid and cell envelope are sorely lacking.
Brian Kloss
New York Structural Biology Center, USA
Title: Structural genomics of integral membrane proteins - past successes and future directions

Biography:
Brian Kloss began his research career as a graduate student in the laboratory of Carter Bancroft at the Mount Sinai School of Medicine, studying the transcriptional regulation of the pituitary-specific prolactin and growth hormone genes. He went on to do a postdoc with Michael Young at Rockefeller University, studying the genetic control of circadian rhythms in Drosophila melanogaster. Afterwards, Brian spent almost six years at a biotech startup, helping to develop a cell-based assay for the screening of ligands of GPCRs. For the past ten years, Brian has been a part of the protein production facility of the Center on Membrane Protein Production and Analysis (COMPPÅ), located at the New York Structural Biology Center. There, he has led a small group focused on the identification, cloning and expression screening of integral membrane proteins of prokaryotic origin, mainly for structural studies.
Abstract:
Approximately one-third of all human genes - as well as genes from most other organisms, across all kingdoms of life - encode integral membrane proteins. Nonetheless, the number of integral membrane protein structures solved lags far behind the number of those solved for their soluble counterparts, due primarily to the difficulty of recombinant expression and the instability of membrane proteins once they are detergent-extracted from the lipid bilayer. Over the past 10-20 years, the number of integral membrane protein structures solved, primarily by X-ray crystallography, has increased significantly and structural genomics approaches have played a considerable role in this progress. More recently, advances in cryo-electron microscopy techniques have permitted structures of integral membrane proteins to be determined at resolutions comparable to that of x-ray crystallography, but requiring much smaller quantities of protein. Concurrently, detergents that improve the stability of integral membrane proteins and purification techniques that allow proteins to be extracted and purified in their native lipid environment have also been developed, allowing structural studies of integral membrane proteins to move forward at an exceedingly rapid pace. I will summarize our past integral membrane protein structural biology efforts that employed structural genomics approaches and high-throughput techniques and describe our plans for future structural studies that will continue to make use genomics-based methods, as well as more recently available reagents, techniques and technologies.
References:
- Su et. al. Structural basis for conductance through TRIC cation channels. Nature Communications 19: 15103 (2017).
- Petrou et. al., Structures of aminoarabinose transferase ArnT suggest a molecular basis for lipid A glycosylation. Science 351: 608-612 (2016).
- Ardiccioni et. al., Structure of the polyisoprenyl-phosphate glycosyltransferase GtrB and insights into the mechanism of catalysis. Nature Communications 7: 10175 (2016).
- Beltrán et. al., Control of carotenoid biosynthesis through a heme-based cis-trans isomerase. Nature Chemical Biology 8: 598-605 (2015).
- Guo et. al., Structure and activity of tryptophan-rich TSPO proteins. Science 347: 551-555 (2015).
Steven Hayward
University of East Anglia, UK
Title: Geometrical Principles of Homeric β-Barrels and β-Helices: Applications to Modelling Amyloid Protofilaments

Biography:
Steven Hayward is Reader in Computational Biology at the University of East Anglia. He uses computational methods, including bioinformatics techniques and simulation, to understand protein structure, dynamics and function. He has focused particularly on protein dynamics and developed the popular DynDom method for the analysis of domain movements in proteins (www.cmp.uea.ac.uk/dyndom). Recently he has worked on the fundamental structure of amyloid fibrils and also works on the development of interactive tools for protein visualization and docking using haptics (www.haptimol.com).
Abstract:
Examples of homomeric β-helices and β-barrels have recently emerged. Here we have generalized the theory for the shear number in β -barrels to encompass β-helices and homomeric structures. We introduce the concept of the “β-strip” which comprises neighboring strands, parallel or antiparallel and forms the repeating unit that builds the helix. In this context the shear number is interpreted as the sum of register shifts between neighboring β-strips. This more general approach has allowed us to derive relationships between the helical width, helical pitch, angle between strand direction and helical axis, mass per length, register shift, and number of strands. The validity and unifying power of the method is demonstrated with known structures including the T4 phage spike, cylindrin, and the HET-s(218-289) prion. The relationships have allowed us to predict register shift and number of strands in transthyretin and Alzheimer β(40) amyloid protofilaments from reported dimensions measured by X-ray fiber diffraction which we have used to construct models that comprise a single strip of in-register β-strands folded into a “β-strip helix”. The results suggest that both stabilization of an individual β-strip helix as a protofilament subunit and growth of the protofilament by the joining of subunits end-to-end, would involve the association of the same pair of sequence segments at the same register shift.

T4 phage spike
References:
- S Hayward, DP Leader, F Alâ€Shubailly, EJ Milnerâ€White (2014) Rings and ribbons in protein structures: Characterization using helical parameters and Ramachandran plots for repeating dipeptides. Proteins 82 (2): 230-239.
- S.Hayward, E.J.Milner-White (2011) Simulation of the β- to α-sheet transition results in a twisted sheet for antiparallel and an α-nanotube for parallel strands: Implications for amyloid formation. Proteins 79(11): 3193-3207.
- D Taylor, G Cawley, S Hayward (2014) Quantitative method for the assignment of hinge and shear mechanism in protein domain movements. Bioinformatics 30(22): 3189-3196.
- S. Hayward and A. Kitao (2015) Monte Carlo Sampling with Linear Inverse-Kinematics for Simulation of Protein Flexible Regions. Journal of Chemical Theory and Computation 11 (8): 3895-3905.
- Georgios Iakovou, Steven Hayward, Stephen Laycock (2017) A virtual environment for studying the docking interactions of rigid biomolecules with haptics. Journal of Chemical Information and Modeling 57 (5): 1142–1152.
Enrica Bordignon
Ruhr-Universität Bochum, Germany
Title: Exploring conformational equilibria of a heterodimeric ABC transporter by electron paramagnetic resonance
Time :
Biography:
Enrica Bordignon is an associate professor at the Ruhr University of Bochum, where she leads an EPR laboratory dedicated to the study of membrane proteins. Approximately 50 papers in protein research by EPR methods. Research interests: understanding the mechanism of action of proteins by EPR.
Abstract:
ABC exporters pump substrates across the membrane by coupling ATP- driven movements of nucleotide binding domains (NBDs) to the transmembrane domains (TMDs), which switch between inward- and outward-facing (IF and OF) orientations. Understanding their cycle has potential for medical applications because they are involved in multidrug resistance of cancer cells.
Site-directed spin labeling electron paramagnetic resonance (EPR), and in particular the dipolar spectroscopy technique called DEER (or PELDOR) was used to investigate the conformational transition of the ABC heterodimeric exporter TM287/288 from the hyperthermophile T. maritima. The analysis revealed that with nucleotides the transporter exists in an equilibrium between the IF and OF states. ATP binding without hydrolysis was sufficient to partially populate the OF state, and an almost complete conformational shift was observed when nucleotides were trapped in a pre- or post-hydrolytic state. At physiological temperature and without nucleotides, the NBDs disengage asymmetrically while the conformation of the TMDs remains unchanged. Nucleotide binding at the degenerate ATP site prevents complete NBD separation, a molecular feature differentiating heterodimeric ABC exporters from their homodimeric counterparts. Our data suggest hydrolysis-independent partial closure of the NBD dimer, which is further stabilized as the consensus site nucleotide is committed to hydrolysis. A unified mechanism is established, which reconciles the available information for heterodimeric ABC exporters1.
References:
- Timachi MH, Hutter CAJ Hohl M, Assafa T, Böhm S, Mittal A, Seeger MA, Bordignon E (2017) Exploring conformational equilibria of a heterodimeric ABC transporter. eLife 6:e20236..
- Celia H, Noinaj N, Zakharov SD, Bordignon E, Botos I, Santamaria M, Barnard TJ, Cramer WA, Lloubes R, Buchanan SK (2016) Structural insight into the role of the Ton complex in energy transduction. Nature 538 (7623):60-65.
- de Almeida Ribeiro E, Pinotsis N, Ghisleni A, Salmazo A, Konarev PV, Kostan J, Sjöblom B, Schreiner C, Polyansky AA, Gkougkoulia EA, Holt MR, Aachmann FL, Žagrović B, Bordignon E, Pirker KF, Svergun DI, Gautel M, Djinović-Carugo K (2014) The structure and regulation of human muscle α-actinin. Cell 159 (6):1447-1460
- Bleicken S, Jeschke G, Stegmueller C, Salvador-Gallego R, García- Sáez AJ, Bordignon E (2014) Structural model of active Bax at the membrane. Molecular Cell 56 (4):496-505.
- Hohl M, Hürlimann LM, Böhm S, Schöppe J, Grütter MG, Bordignon E, Seeger MA (2014) Structural basis for allosteric cross-talk between the asymmetric nucleotide binding sites of a heterodimeric ABC exporter. PNAS 111 (30):11025-11030.
J. Chakrabarti
S. N. Bose National Centre for Basic Sciences, India
Title: Microscopic calculation of conformational thermodynamics in bio-macromolecular complexes
Biography:
Jaydeb Chakrabarti is a theoretical physicist trained in condensed matter physics. He looks into Statics and Dynamics of soft matter systems including bio-macromolecules. The theoretical methods used in these studies are: (1) Computer simulations based on Molecular Dynamics, Monte Carlo and Brownian Dynamics; (2) Mean field calculations based on classical density functional theories. The goal of his researches is to relate macroscopic properties to microscopic motions.
Abstract:
Statement of the Problem: The microscopic basis of connection between protein conformation and function is a fundamental challenge. Recent experiments show the importance of conformational changes in providing stability to protein complexes. The changes in conformational state have both enthalpy and entropic components. It has been possible to quantify the entropy changes due conformational changes from Nuclear magnetic Resonance data. Here we show microscopic calculation of both the enthalpy and entropy contribution while protein conformations change from the equilibrium distribution of the dihedral angles of proteins. We have shown that the free-energetically destabilized and entropically disordered residues in a given conformation compared to a reference conformation act as binding residue in the giv en conformation. Here we show that this principle can: (1) ascertain ligand binding of a protein in different conformations; (2) supplement the structure of missing fragments in a protein; and (3) serve as a guideline for allosteric changes. All these applications point to tune protein in-silico which would help to design functional materials with protein as building blocks.
References:
- Conformational thermodynamics guided structural reconstruction of biomolcular fragments, Samapan Sikdar, J. Chakrabarti and Mahua Ghosh, Molecular Biosystems, 12, 444-453 (2016).
- A microscopic insight from conformational thermodynamics to functional ligand binding in proteins, Samapan Sikdar, J. Chakrabarti and Mahua Ghosh, Molecular Biosystems, 10, 3280-3289 (2014).
- Quantum chemical studies on the role of residues in calcium binding to Calmodulin, Samapan Sikdar, Mahua Ghosh, Molly De Raychaudhury and J. Chakrabarti, Chem Phys Lett, 605-606, 103-107(2014).
- Conformational contribution to thermodynamics of binding in protein-peptide complexes through microscopic simulation Amit Das, J. Chakrabarti and Mahua Ghosh, Biophysical Journal, 104, 1274 (2013).
- High-Affinity Quasi-Specific Sites in the Genome: How the DNA-Binding Proteins Cope with them J. Chakrabarti, Navin Chandra, Paromita Raha, and Siddhartha Roy, Biophysical Journal, 101, 1123 (2011).
- Track 10: Signalling Biology
Session Introduction
Irena Levitan
University of Illinois at Urbana–Champaign, USA
Title: Structural insights into cholesterol regulation of inwardly-rectifying K+ channels

Biography:
Irena Levitan has done her PhD and is a Professor of Medicine and Adjunct Professor of Bioengineering at the University of Illinois at Chicago. Her current research focuses on cholesterol regulation of ion channels and cellular biomechanics. Her group has provided the first comprehensive structural insights into cholesterol regulation of K+ channels and the cross-talk between cholesterol and other regulators of these channels. She was named a Guyton Distinguished Lecturer by the Association Chairs of Departments of Physiology for her quantitative and biophysical work on cholesterol modulation of ion channels and how this can affect integrated organ function. She is an author of more than 70 publications and a leading Editor of Cholesterol Regulation of Ion Channels and Receptors (Wiley, 2012) and Vascular Ion Channels in Physiology and Disease (Springer, 2016).
Abstract:
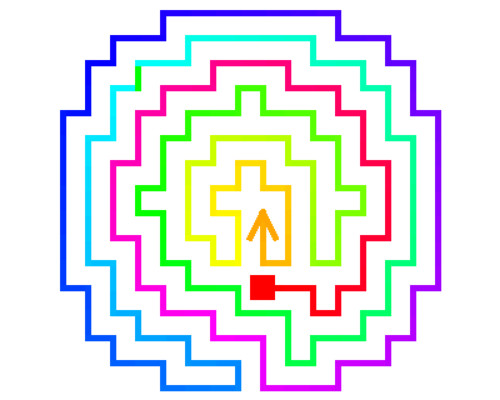
Cholesterol is known to play a major role in regulating the function of multiple membrane proteins including a growing number of ion channels. Our studies focus on inwardly-rectifying K+ (Kir) channels that are ubiquitously expressed in mammalian cells and are known to play major role in membrane excitability and shear stress sensation. In this study, we have shown that Kir channels are suppressed by loading the cells with cholesterol and enhanced by cholesterol depletion. A series of studies revealed that cholesterol interacts with the channels directly by stabilizing them in a long-lived closed “silent” state and that multiple structural features of the channels are essential for conferring their cholesterol sensitivity. Using a combined computational-experimental approach, we show that cholesterol may bind to two nonannular regions that form hydrophobic pockets between the transmembrane helices of the adjacent subunits of the channel. The location of the binding regions suggests that, cholesterol modulates channel function by affecting the hinging motion at the center of the porelining transmembrane helix that underlies channel gating. In addition, we identified a series of residues in the C and N-terminus of the channel. These are critical for conferring cholesterol sensitivity to the channels, but are not part of the binding sites. These residues form a distinct cytosolic structure, a cholesterol sensitivity belt which surrounds the cytosolic pore of the channel in proximity to the transmembrane (TM) domain, and includes residues whose mutation results in abrogation of the channel’s cholesterol sensitivity. Further analysis identified a reversal residue chain comprised of residues that link one of the cholesterol sensitivity belt residues with a distant cytosolic residue that constitute a two-way molecular switch of the channel sensitivity to cholesterol. Further studies are needed to elucidate the connection between cholesterol binding and channel.
References:
- Rosenhouse-Dantsker A, Noskov S, Durdagi S, Logothetis DE, Levitan I. Identification of novel cholesterol-binding regions in Kir2 channels. JBC, 288(43):31154-64, 2013
- Han H., Rosenhouse-Dantsker A. Gnanasambandam R. Epshtein Y. Chen Z., Sachs F, Minshall RD and I Levitan. Silencing of Kir2 channels by caveolin-1: cross-talk with cholesterol. Journal of Physiology, 592:4025-38, 2014
- Rosenhouse-Dantsker A. Epshtein Y. and I. Levitan. Interplay between lipid modulators of Kir2 channels: cholesterol and PIP2. Computational and Structural Biotech Journal, 11(19):131-7, 2014
- Bukiya AN, Osborn CV, Kuntamallappanavar G, Toth PT, Baki L, Kowalsky GB, Oh MO, Dopico AM, Levitan I. and A. Rosenhouse-Dantsker. Cholesterol increases the open probability of cardiac KACh currents, BBA Membranes, 848:2406-13, 2015
- Ahn SJ, Fancher IS, Bian JT, Zhang CX, Schwab S, Gaffin R, Phillips SA, and I Levitan. Inwardly-rectifying K+ channels are major contributors to flow-induced vasodilation in resistance arteries. J of Physiology. In Press.
Joachim Krebs
Max Planck Institute for Biophysical Chemistry, Germany
Title: Calcium, Calmodulin and the Plasma Membrane Calcium Pump

Biography:
Prof. RJP Williams at Oxford, UK. In 1977 he accepted a staff position at the Institute of Biochemistry at the Swiss Federal Institute of Technology (ETH) in Zürich, Switzerland. He was lecturing different courses in biochemistry and biophysics, and was leading a Lab working on the structure-function relationship of calcium-binding and calcium-transporting proteins. After retirement from the ETH he continued his research as a consultant of the Lab of Prof. Christian Griesinger at the MPI in Göttingen, Germany. He has authored, co-authored and edited numerous articles in international journals. Recently he edited a book on “Calcium: A matter of life or death” published in 2007. He is at the Editorial Board of BBA Molecular Cell Research and Archives of Biochemistry and Biophysics.
Abstract:
Calcium is the third most abundant metal in nature and a versatile carrier of many signals within and outside the cell. Due to its peculiar coordination chemistry calcium is highly flexible as a ligand which enables it to regulate many important aspects of cellular activity. Calcium can fulfill its many different functions insite and out of the cell due to an integrated network of calcium channels, exchangers and pumps. In this presentation I will give an overview on our studies of calcium binding proteins, their interaction with protein targets resulting in specific modulations of protein-protein interactions. This will be demonstrated by the interaction of the calcium binding protein calmodulin with one of its targets, the plasma membrane calcium pump, an important regulator of calcium homeostasis of the cell.
References:
- Krebs, J. (2015) The plethora of PMCA isoforms: Alternative splicing and differential expression. Biochim. Biophys. Acta 1853: 2018-2024
- Seidel, K. et al. (2008) Structural characterization of Ca(2+)-ATPase-bound phospholamban in lipid bilayers by solid-state nuclear magnetic resonance (NMR) spectroscopy. Biochemistry 47: 4369-4376.
- Elshorst, B., Krebs, J. et al. (1999) NMR solution structure of a complex of calmodulin with a binding peptide of the Ca2+-pump. Biochemistry 38: 12320-12332.
- Carafoli, E., Krebs, J. (2016) Why Calcium? How Calcium became the best Communicator. J. Biol. Chem. 291: 20849-20857.
5.Toyoshima, C. (2009) How Ca2+-ATPase pumps ions across the sarcoplasmic reticulum membrane. Biochim. Biophys. Acta 1793: 941-946.
Carol A. Heckman
Bowling Green State University, USA
Title: Structural aspects of cell signaling
Time :

Biography:
Carol Heckman is an expert on preneoplasia and image analysis and has developed optical and computational methods suitable for cell feature analysis. Applying these methods, she showed how to deconstruct the cell phenotype and find the features related to oncogenic transformation. About half of the 20 features are useful in distinguishing the phenotypes of normal and cancerous epithelial cells. She has published numerous papers on these topics. The cell features form the basis of an assay to flag chemicals of interest for drug development and identify diseases that can be targeted productively by existing drugs.
Abstract:
Statement of the problem: Endpoints such as adhesion and motility have been used to infer the function of a protein in cells. These endpoints are unsatisfactory, because a protein can be recruited to different substructures and promote different outcomes in such structures. By defining meaningful endpoints, it is possible to identify a protein’s contribution to several different patterns of cell organization and thereby address major problems in biology.
Methodology and theoretical orientation: We developed an unbiased method of classifying and quantifying features of fixed, adherent epithelial cells. Primary data, consisting of 102 measures of contour geometry, curvature, relationship to derived model figures, etc., were used to calculate 20 latent factors representing cell features. Factors detect structure by recognizing the relationships between variables. Cells from experiments are classified according to each factor by summing the factor loadings. Filopodia (factor 4) accounted for a larger proportion of cancer-related variance than any other feature. Filopodia are the sensory appendages that are relied on when cells distinguish their more and less adhesive sides. The protrusion defined as factor 7 represented neurites. Even when small, neurites differed from lamellipodia (factor 5). Several factors contribute to ruffling.
Findings: Filopodia are down-regulated by three isoforms of protein kinase C (PKC). The effect of PKC ε, a known oncogene, on filopodia is only observed after tumor promoter treatment. The effect is in part due to a PKC ε-mediated increase in ruffling. In cells not treated with tumor promoter, filopodia are down-regulated by isoforms a and h. PKC a has contrary effects in promoter-treated cells, where it conserves filopodia by suppressing ruffling activity. Activated PKC a may promote filopodia. These activities are consistent with the concept that filopodia are implicated in cell homeostasis. By regulating the prevalence of filopodia, PKC can regulate the way cells react to their surroundings.
References:
- Heckman CA, Pandey P, Cayer ML, Biswas T, Zhang ZY, Boudreau NS (2017) The tumor promoter-activated protein kinase Cs are a system for regulating filopodia. Cytoskeleton (Hoboken) May 8. doi: 10.1002/cm.21373. [Epub ahead of print]
- Mukhopadhyay C, Triplett A, Bargar T, Heckman C, Wagner K-, M Naramura M (2016) Casitas B-cell lymphoma (Cbl) proteins protect mammary epithelial cells from proteotoxicity of active c-Src accumulation. PNAS USA 113: E8228-E8237
- Amarachintha SP, Ryan KJ, Cayer M, Boudreau NS, Heckman CA (2014) Effect of Cdc42 domains on filopodia sensing, cell orientation, and haptotaxis. Cellular Signalling S0898-6568(14)00379-9. doi: 10.1016/j.cellsig.2014.11.025.
- Heckman CA, Plummer HK III (2013) Filopodia as sensors. Cellular Signaling 25: 2298-2311. doi: 10.1016/j.cellsig.2013.07.006.
- Heckman CA, Varghese M, Cayer ML, Boudreau NS (2012) Origin of ruffles: Linkage to other protrusions, filopodia and lamellae. Cellular Signaling 24: 189-198.
Vesa P Hytönen
University of Tampere, Finland
Title: Mechanical Stability of Talin Rod Controls Traction Force Generation and Cell Migration

Biography:
Vesa P Hytönen is a Head of the Protein Dynamics research group in BioMediTech at the University of Tampere, Tampere, Finland. After graduating as a PhD from the University of Jyväskylä, Jyväskylä, Finland in 2005, he has conducted Post-doctoral training at ETH Zurich, Zürich, Switzerland from 2005-2007. He then continued as a Post-doctoral researcher at the University of Tampere and established independent research group in 2010. He is currently working as Associate Professor at the University of Tampere. His research interests are mechanobiology, protein engineering and vaccine research and authored more than 100 scientific articles.
Abstract:
Talin is a central adhesion protein linking β-integrin cytosolic domains to actin fibers. It participates in the transmission of mechanical signals between extracellular matrix and cell cytoskeleton. Talin rod domain consists of a series of mechanically vulnerable α-helical subdomains containing binding sites for other adhesion proteins such as vinculin, actin and RIAM. Force induced unfolding of these rod subdomains has been proposed to act as a cellular mechanosensor, but so far evidence linking their mechanical stability and cellular response has been lacking. We show that stepwise mechanical destabilization of talin rod subdomain increases talin and vinculin accumulation into cell-matrix adhesions and decreases cell migration rate. In addition, mechanical destabilization of talin subdomain was found to decrease cellular traction force generation and to promote the formation of adhesions on fibronectin over vitronectin. Experiments with truncated talin forms confirmed the mechanosensory role of the talin subdomain and excluded the possibility that the observed effects are caused solely by the release of talin autoinhibition. We demonstrate that by modulating the mechanical stability of an individual talin rod sub-domain, it is possible to affect traction force generation, ECM sensing and consequently highly coordinated processes such as cell migration. Our results suggest that talin acts as a mechanosensor and is responsible for controlling the cellular processes dependent on mechanical signals and cellular mechanosensing.
References:
- von Essen M, Rahikainen R, Oksala N, Raitoharju E, Seppälä I, Mennander A, Sioris T, Kholová I, Klopp N, Illig T, Karhunen PJ, Kähönen M, Lehtimäki T, Hytönen VP (2016) Talin and vinculin are downregulated in atherosclerotic plaque; Tampere Vascular Study. Atherosclerosis. 255:43-53.
- Qi L, Jafari N, Li X, Chen Z, Li L, Hytönen VP, Goult BT, Zhan CG, Huang C (2016) Talin2-mediated traction force drives matrix degradation and cell invasion. J Cell Sci. 129:3661-3674.
- Haining AW, von Essen M, Attwood SJ, Hytönen VP, Del Río Hernández A (2016) All Subdomains of the Talin Rod Are Mechanically Vulnerable and May Contribute To Cellular Mechanosensing. ACS Nano 10:6648-58.
- Hytönen VP, Wehrle-Haller B (2016) Mechanosensing in cell-matrix adhesions - Converting tension into chemical signals. Exp Cell Res. 343:35-41.
- Hytönen VP, Vogel V (2008) How force might activate talin's vinculin binding sites: SMD reveals a structural mechanism. PLoS Comput Biol. 4:e24.
Biography:
Francis Millett received his B.S in Chemistry from the University of Wisconsin in 1965, his Ph.D. in Chemical Physics from Columbia University in 1970, and was an NIH Postdoctoral Fellow at California Institute of Technology from 1970-1972. He joined the faculty of the University of Arkansas in 1972, and is now a Distinguished Professor. He developed, together with Bill Durham, the ruthenium photoreduction method which made it possible to measure the kinetics of key steps in electron transfer during mitochondrial oxidative phosphorylation. He has directed collaborative, multidisciplinary research which combines rapid kinetics methods, site-directed mutagenesis, X-ray crystallography, and NMR to investigate protein structure-function relationships.
Abstract:
The electron transfer reactions within wild-type Rhodobacter sphaeroides cytochrome bc1 (cyt bc1) were studied using a ruthenium dimer to rapidly photooxidize cyt c1. It was found that when cyt bH was initially reduced before the reaction, photooxidation of cyt c1 led to bifurcated reduction of both the iron-sulfur protein and cyt bL by QH2 in the Qo site, followed by re-oxidation of two equivalents of cyt bL and cyt bH. It was proposed that the newly formed ubiquinone diffused through the hydrophobic cavity linking the Qo site of the reactive monomer A to the Qi site of the other monomer B, leading to oxidation of cyt bH in monomer B followed by oxidation of cyt bL in monomer A by cross-monomer electron transfer. Addition of one equivalent of the Qi site inhibitor antimycin to the cyt bc1 dimer had very little effect on any of the electron transfer reactions, while addition of a second equivalent completely inhibited re-oxidation of cyt bL and cyt bH. It was also found that addition of one equivalent of the Qo site inhibitor stigmatellin to the cyt bc1 dimer completely inhibited all electron transfer reactions in both monomers of the dimer. These experiments are consistent with a half-of-the-sites mechanism in which only one monomer of the dimer is active at a time, implying monomer-monomer interactions. The rapid electron transfer reaction from the ISP to cyt c1 was found to be greatly decreased by viscosity, indicating a multi-step diffusional mechanism as the iron-sulfur protein rotates from the b state to the c1 state.
References:
- Janzon, Julia; Yuan, Quan; Malatesta, Francesco; Hellwig, Petra; Ludwig, Bernd; Durham, Bill; Millett, Francis. “Probing the Paracoccus denitrificans cytochrome c1-Cytochrome c552 interaction by mutagenesis and fast kinetics,” Biochemistry (2008) 47(49), 12974-12984.
- Mills, D.Z.; Xu, Shujuan; Geren, L.; Hiser, C.; Qin, L.; Sharpe, M.A.; McCracken, J.; Durham, B.; Millett, F.; Ferguson-Miller, S. Proton-Dependent Electron Transfer from CuA to Heme a and Altered EPR Spectra in Mutants Close to Heme a of Cytochrome Oxidase. Biochemistry (2008), 47(44), 11499-11509.
- Castellani, M.; Havens, J.; Kleinschroth, T.; Millett, F.; Durham, B.; Malatesta, F.; Ludwig, B. “The acidic domain of cytochrome c(1) in Paracoccus denitrificans, analogous to the acidic subunits in eukaryotic bc(1) conmplexes, is not involved in the electron transfer reaction to its native substrate cytochrome c(552). Biochim Biophys Acta (2011), 1807:1383-1389
- Havens, J.; Castellani, M.; Kleinschroth, T.; Ludwig, B.; Durham, B.; Millett, F. Photoinitiated electron transfer within the paracoccus denitrificans cytochrome bc(1) complex: Mobility of the iron-sulfur protein is modulated by the occupant of the Q(o) site. Biochemistry (2011) Dec 6;50(48):10462-10472.
- Durham B, Millett F. “Design of photoactive ruthenium complexes to study electron transfer and proton pumping in cytochrome oxidase.” Biochim Biophys Acta. (2012) Apr;1817(4):567-74.
Leonas Valkunas
Vilnius university, Lithuania
Title: Mapping energy transfer channels in fucoxanthin–chlorophyll protein complex

Biography:
Leonas Valkunas is the author of more than 350 international publications, head of the department of Theoretical Physics at Vilnius university and head of the Department of Molecular Compounds Physics at the Center for Physical Sciences and Technology in Vilnius. He is involved in studies of primary processes of photosynthesis such as excitation dynamics and photoinduced charge separation in various photosynthetic systems based on the spectroscopic data and using various theoretical modelling approaches.
Abstract:
Fucoxanthin–chlorophyll protein (FCP) is the key molecular complex performing the light-harvesting function in diatoms, which, being a major group of algae, are responsible for up to one quarter of the total primary production on Earth. These photosynthetic organisms contain an unusually large amount of the carotenoid fucoxanthin, which absorbs the light in the blue–green spectral region and transfers the captured excitation energy to the FCP bound chlorophylls. Due to the large number of fucoxanthins, the excitation energy transfer cascades in these complexes are particularly tangled. Energy transfer processes and coherent phenomena in the fucoxanthin–chlorophyll protein complex, which is responsible for the light harvesting function in marine algae diatoms, were investigated at 77 K by using two-dimensional electronic spectroscopy. Experiments performed on femtosecond and picosecond timescales led to separation of spectral dynamics, witnessing evolutions of coherence and population states of the system in the spectral region of Qy transitions of chlorophylls a and c. Analysis of the coherence dynamics allowed us to identify chlorophyll (Chl) a and fucoxanthin intramolecular vibrations dominating over the first few picoseconds. Closer inspection of the spectral region of the Qy transition of Chl c revealed previously not identified, mutually non-interacting chlorophyll c states participating in femtosecond or picosecond energy transfer to the Chl a molecules. Consideration of separated coherent and incoherent dynamics allowed us to hypothesize the vibrations-assisted coherent energy transfer between Chl c and Chl a and the overall spatial arrangement of chlorophyll molecules.
References:
- Butkus V, Gelzinis A, Augulis R, Gall, A, Büchel C, Robert B, Zigmantas D, Valkunas L, Abramavicius D (2015) Coherence and population dynamics of chlorophyll excitation in FCP complex: two-dimensional spectroscopy study. J. Chem. Phys. 142: 212414.
- Gelzinis A, Butkus V, Songaila E, Augulis R, Gall A, Büchel C, Robert B, Abramavicius D, Zigmantas D, Valkunas L (2015) Mapping energy transfer channels in fucoxanthin-chlorophyll protein complex. Biochim. Biophys. Acta 1847: 241-247.
- Farooq S, Chmeliov J, Trinkunas G, Valkunas L, van Amerongen H. (2016) Is there excitation energy transfer between different layers of stacked photosystem II containing thylakoid membranes? J. Phys. Chem. Lett.7: 1406-1410.
- Chmeliov J, Gelzinis A, Songaila E, Augulis R, Duffy CDP, Ruban AV, Valkunas L. (2016) The nature of self-regulation in photosynthetic light-harvesting antenna. Nature Plants 2: 16045.
- Abramavicius D, Valkunas L. (2016) Role of coherent vibrations in energy transfer and conversion in photosynthetic pigment-protein complexes. Photosynth. Res. 127: 33-47.

Biography:
Jianmin Cui is a professor on the Spencer T. Olin Endowment at Washington University in St. Louis, in the Department of Biomedical Engineering. He received Ph.D. in Physiology and Biophysics from State University of New York at Stony Brook and a post-doctoral training at Stanford University. He was an assistant professor of Biomedical Engineering at Case Western Reserve University before moving to St. Louis. His research interests include BK-type calcium-activated potassium channels and IKs channels.
Abstract:
Voltage-gated ion channels generate dynamic ionic currents that are vital to the physiological functions of many tissues. These proteins contain separate voltage-sensing domains, which detect changes in transmembrane voltage, and pore domains, which conduct ions. Coupling of voltage sensing and pore opening is critical to the channel function and has been modeled as a protein–protein interaction between the two domains. However, our data show that coupling in Kv7.1 channels requires the lipid phosphatidylinositol 4,5-bisphosphate (PIP2). We found that voltage-sensing domain activation failed to open the pore in the absence of PIP2. This result is due to loss of coupling because PIP2 was also required for pore opening to affect voltage-sensing domain activation. We identified a critical site for PIP2-dependent coupling at the interface between the voltage-sensing domain and the pore domain. This site is actually a conserved lipid-binding site among different K+ channels, suggesting that lipids play an important role in coupling in many ion channels. To further investigate the mechanism of PIP2 mediated VSD-pore coupling, we identified a compound that mimics PIP2 structure and function as a molecular probe. This compound was identified using an in silico screening approach based on molecular docking of a library of compounds to the PIP2 binding site in a homology model of the Kv7.1 channel. Our results show that this compound can substitute PIP2 in activating the Kv7.1 channel.
References:
- Cui, J. (2016) Voltage dependent gating: novel insights from KCNQ1 channels. Biophys J 110:14-25.
- Kasimova M, Zaydman M, Cui J, Tarek M (2015) PIP2-dependent coupling is prominent in Kv7.1 due to weakened S4-S5/S6 interactions. Scientific Reports 5:7474.
- Zaydman, M.A. and Cui, J. (2014) PIP2 activation of KCNQ channels. Front Physiol. 5:195. doi: 10.3389/fphys.2014.00195.
- Zaydman M, Kasimova M, McFarland K, Liang H, Beller Z, Shi J, Hou P, Kinser H, Zhang G, Tarek M, and Cui J (2014) Domain-domain interactions determine the gating, permeation, pharmacology, and subunit modulation of the IKs ion channel. eLife 2014;3:e03606.
- Zaydman MA, Silva JR, Delaloye K, Li Y, Liang H, Larsson HP, Shi J, and Cui J (2013) Kv7.1 ion channels require PIP2 to couple voltage sensing to pore opening. Proc. Natl. Acad. Sci., U.S.A. 110:13180-13185.
Igor L. Barsukov
University of Liverpool, UK
Title: LD Motif Interacting networks in cell-matrix adhesion.
Time :

Biography:
Igor Barsukov has expertise in structural biology, primarily using NMR spectroscopy and X-ray crystallography. The main focus of his research has been on the structure and function of integrin-mediated cell-matrix adhesions, where he directed full structural analysis of the key adhesion proteins talin, leading to the currently widely used model of stretch-dependent talin activation. He is currently extending the model of talin functionality to include competitive, talin based, interaction networks. Recently he identified interactions between neuronal scaffold proteins Shank3 and Ras-family GTPase that regulate integrin activity and have implications for control of synaptic plasticity.
Abstract:
Statement of the Problem: Cell-matrix adhesion requires assembly of large multi-protein complexes linked to the cyto-domains on the integrin adhesion receptors. These complexes dynamically change in response to the adhesion forces and environmental signals. Mechano-sensitive adaptor protein talin couples the force and adhesion signaling by acting as a hub for numerous, often competitive, interactions. The molecular mechanism that controls the co-ordination of the interactions in time and space is currently not understood. The aim of this study is to define the interactions between talin and Rho-GAP Deleted in Liver Cancer 1 (DLC1) that regulates adhesion forces. Methodology: Structure of the talin/DLC1 complex was solved by X-ray crystallography and the interactions between the proteins analysed by NMR spectroscopy. Fluorescent imaging was used to define the interactions within the adhesion complexes and cancer cell lines were employed to characterise the effect of the interaction on the biological activity. Findings: We defined the atomic details of the talin/DLC1 interactions and used this information to identify signalling protein paxillin as a talin ligand. Based on the structural information we designed a range of talin mutants that modulate the interactions and demonstrated that the mutations reduce DLC1 signalling in adhesion. Conclusion & Significance: Talin recognises LD motifs in DLC1 and paxillin through a set of well-defined charge interactions. These interactions are similar to other LD motif interactions previously identified in signalling pathways. These interactions are also similar to the interactions between talin, RIAM and vinculin that were previously not assigned to the LD motif family. Together, the relatively weak LD motif interactions within the adhesion complex create a protein network that could dynamically respond to the adhesion signals.
References:
- Zacharchenko, T., et al., LD Motif Recognition by Talin: Structure of the Talin-DLC1 Complex. Structure, 2016.
- Atherton, P., et al., Vinculin controls talin engagement with the actomyosin machinery. Nat Commun, 2015. 6: p. 10038.
- Goult, B.T., et al., RIAM and Vinculin Binding to Talin Are Mutually Exclusive and Regulate Adhesion Assembly and Turnover. J Biol Chem, 2013. 288(12): p. 8238-8249.
- Goult, B.T., et al., Structural studies on full-length talin1 reveal a compact auto-inhibited dimer: Implications for talin activation. J Struct Biol, 2013. 184(1): p. 21-32.
- Elliott, P.R., et al., The Structure of the Talin Head Reveals a Novel Extended Conformation of the FERM Domain. Structure, 2010. 18(10): p. 1289-1299.
Arfaxad Reyes Alcaraz
Korea University College of Medicine, South Korea
Title: Structural Conformational changes report biased agonism: The case of Galanin receptors

Biography:
Arfaxad Reyes has his expertise in structure and stability of G-protein coupled receptors and passion for improving and creating new drug discovery platforms that greatly contribute in the development of more selective drugs with minor side effects. His studies about biased agonism in Galanin receptors help to understand the relationship between conformational structure of the receptor and its corresponding physiological effect induced by a specific ligand. Recently he and co-workers were able to develop a highly selective agonist for Galanin receptor 2 with anxiolytic effect in vivo (Arfaxad Reyes-Alcaraz et al Sci. Rep 2016) which was the base to discover how different ligand structures induce different conformations on the structure of Galanin receptors. This work greatly contribute to understand the relationship between structure and function of Galanin receptors.
Abstract:
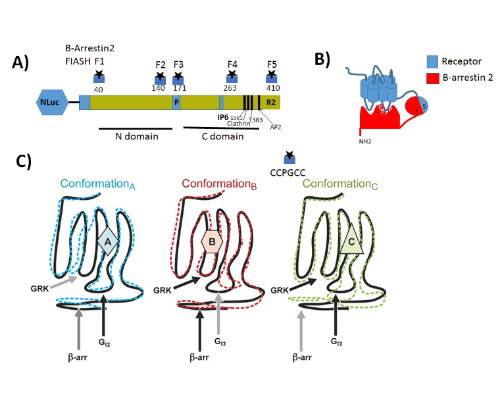
Statement of the Problem: G protein coupled receptors (GPCRs) also known as seven-transmembrane receptors and are the largest family of cell-surface receptors that communicate extracellular stimuli to the cell interior (1). It is now accepted that chemically distinct ligands bind to the same GPCR and can stabilize the receptor in multiple active conformations, which results in differential activation of cell signaling pathways and, eventually, in different physiologic outcomes a phenomenon known as biased agonism (2). Biased agonism can be exploited to design drugs that selectively activate signaling pathways, leading to the desired physiologic effects while on target side effects elicited by activation of other signaling pathways via the same receptor subtype (3). Methodology & Theoretical Orientation: The aim of this study was to stablish a relationship between conformational changes in Galanin receptors and their signaling properties in living cells, for that purpose we develop a structural complementation assay based on NanoBit technology and a series of conformational fluorescein arsenical hairpin (FIASH) bioluminescence resonance energy transfer (BRET) biosensors to monitor structural changes β-arrestin 2 induced by the binding with each Galanin receptor. Findings: Here we show that Galanin receptors impose different conformational signatures in β-arrestin, moreover structurally different ligands activating the same receptor imposed different conformations in β-arrestin 2 producing biased signaling. Conclusion & Significance: Our data provide definite evidence that a receptor activated by structurally different ligands can adopt multiple active conformations. Moreover, this finding also demonstrates that functionally specific structural Galanin receptor conformations can indeed be translated to downstream effectors producing a different physiological response.
References:
- Reyes-Alcaraz A., Lee, Y. N., Son, G. H., Kim, N. H., Kim, D. K., and Yun, S., Kim D. H., Hwang, J. I. and Seong, J. Y. Development of Spexin-based Human Galanin Receptor Type IISpecific Agonists with Increased Stability in Serum and Anxiolytic Effect in Mice. Scientific Reports, 6:21453 (2016).
- Reyes-Alcaraz A., Matinez-Archundia M., Ramon E. and Garriga P. Salt Effects on the Conformational Stability of the Visual G-Protein-Coupled Receptor Rhodopsin. Biophysical Journal, 101, 2798-2806 (2011).
- Moon M. J., Lee Y.-N., Park S., Reyes-Alcaraz A., Hwang J. –I., Millar R. P., Choe H. and Seong J. Y. Ligand Binding Pocket Formed by Evolutionarily Conserved Residues in the Glucagon-like Peptide-1 (GLP-1) Receptor Core Domain. The Journal of Biological Chemistry, 290, 5696-5706, (2015).
- Yun, S., Furlong M., Sim M., Cho M., Park, S., Cho E. B., Reyes-Alcaraz, A., Hwang, J. -I., Kim, J., and Seong J. Y. Prevertebrate Local Gene Duplication Facilitated Expansion of the Neuropeptide GPCR Superfamily. Molecular Biology and Evolution 1-15, (2015).
- Reyes-Alcaraz A., Tzanov T. and Garriga P. Stabilization of Membrane Proteins: the Case of G-Protein-Coupled Receptors. Engineering in Life Sciences, 8, 207–217 (2008).
Alessandra Astegno
University of Verona, Italy
Title: A biophysical and structural approach to investigate calcium sensor properties of plant calmodulin-like proteins
Biography:
Alessandra Astegno is interested in different aspects of protein chemistry, including folding, evolution and structure-function relationship of proteins and macromolecular assemblies. She obtained a PhD in Applied Biotechnologies from University of Verona in 2010. She is currently assistant professor in Biochemistry at the Department of Biotechnology of the University of Verona. She has a solid background in recombinant protein expression and purification, functional and structural characterization of metallo-proteins as well as PLP-dependent enzymes. Recently, her work focused on the study of calcium signaling in higher plants through biophysical, biochemical and structural characterization of calcium sensor proteins, such as calmodulin and calmodulin like proteins of Arabidopsis thaliana.
Abstract:
Calcium is an essential second messenger in plants that regulates various signaling pathways through stimulus-specific Ca2+ signatures, which are decoded and converted into a wide variety of biochemical changes by Ca2+ sensors. Besides evolutionarily conserved calmodulin (CaM), plants exclusively possess a group of calmodulin-like proteins (CMLs), which play central roles in the coordination of plant responses to different external stimuli. Nevertheless, only few of these proteins have been thoroughly characterized and demonstrated to function as Ca2+ sensors.
Our research is focused on the investigation of the metal-binding, physicochemical and structural properties of various plant CMLs using complementary biophysical and structural approaches in order to correlate their properties with the biological activity.
We have recently characterized CML36 from Arabidopsis thaliana, demonstrating that in vitro the protein shows features consistent with Ca2+ sensor function. ITC analysis revealed that CML36 possesses two high affinity Ca2+/Mg2+ mixed sites and two low affinity Ca2+-specific sites. Binding of Ca2+ to CML36 increases its α-helical content and triggers a conformational change that exposes hydrophobic surfaces necessary for target recognition. Ca2+ and Mg2+ ions also stabilized the tertiary structure of CML36. In particular, cations binding to the Ca2+/Mg2+ mixed sites appears to guide a large structural transition from a loosely packed molten globule apo-state to a well-defined, stable holo-structure. Through in vitro binding experiments, we showed that CML36 directly interacts with the N-terminal domain of Arabidopsis Ca2+-ATPase isoform 8 (ACA8), a type IIB Ca2+ pump localized at the plasma membrane (PM). Moreover, we demonstrated that this interaction promotes ACA8 Ca2+-dependent hydrolytic activity in vitro.
References:
- Astegno A, Maresi E, Bertoldi M, La Verde V, Paiardini A, Dominici P (2017) Unique substrate specificity of ornithine aminotransferase from Toxoplasma gondii. Biochem J. 474(6):939-955.
- Vallone R, La Verde V, D'Onofrio M, Giorgetti A, Dominici P, Astegno A (2016) Metal binding affinity and structural properties of calmodulin-like protein 14 from Arabidopsis thaliana. Protein Sci. 25(8):1461-71.
- Kumar N, Astegno A, Chen J, Giorgetti A, Dominici P (2016) Residues in the distal heme pocket of Arabidopsis non-symbiotic hemoglobins: implication for nitrite reductase activity. Int J Mol Sci. 28;17(5).
- Astegno A, La Verde V, Marino V, Dell'Orco D, Dominici P (2016) Biochemical and biophysical characterization of a plant calmodulin: Role of the N- and C-lobes in calcium binding, conformational change, and target interaction. Biochim Biophys Acta. 1864(3):297-307.
- Astegno A, Capitani G, Dominici P (2015) Functional roles of the hexamer organization of plant glutamate decarboxylase. Biochim Biophys Acta. 1854(9):1229-37.
He Jianwei
Liaoning University, China
Title: Structural insights into the mechanism of how polyphenols suppresses amyloid fibrillation
Biography:
He Jianwei, professor at School of Life Science, Liaoning University, P.R.China, received M.S. degrees in Biochemistry in 2002, from Yamaguchi University, Japan. He completed his Ph.D in Bioresource in 2005 at Tottori University, Japan, his research interests include: 1) Using molecular dynamics and biochemical methods to study protein oligomerization progress and the importance of dimers and tetramers in the aetiology of amyloidotic diseases. 2) Mining, screening or designing of novel inhibitors of natural resources against protein misfolding and amyloid aggregation.
Abstract:
Polyphenols, especially natural flavonols, have received considerable public attention in China due to the positive association between food and traditional herbal consumption and beneficial health effects. Flavonoids has been demonstrated to be active inhibitors of fibrillation by amyloidogenic protein. We recently reported the inhibitory activity of Myricetin against HEWL fibril formation, in which Myricetin exhibited a stronger inhibition than the well-characterized polyphenol Quercetin. In contrast to our previous studies using other polyphenols, we find the generation of irregular structural aggregates formed by the binding of Morin to HEWL, which support a novel and distinctive model for how this small molecule inhibits amyloid formation. Moreover, we also demonstrated that EGCG was a potent inhibitor of amyloidogenic cystatin amyloid fibril formation in vitro. Through combining experimental and computational data, we were able to propose a mechanism by which EGCG inhibited the fibrillation of cystatin: EGCG appears to be a generic inhibitor of amyloid-fibril formation, although the mechanism by which it achieves such inhibition may be specific to the target fibril-forming polypeptide. In conclusion, our findings implicate the importance of diet and drink habits as playing a major role in guarding against amyloid fibril formation and promoting healthy aging.

References:
- Chong Xiaoying, Sun Luchen, Sun Yonghui, Chang Lin, Chang K Alan, Lu Xian, Zhou Xuejie, Liu Junqing, Zhang Bing, Jones W Gary, He Jianwei(*). Insights into the mechanism of how Morin suppresses amyloid fibrillation of hen egg white lysozyme. Int J Biol Macromol. 2017, 101: 321-325.
- Wang Na., He Jianwei(*), Chang Alan K., Wang Yu, Xu Linan, Chong Xiaoying, Lu Xian, Sun Yonghui, Xia Xichun, Li Hui, Zhang Bing, Song Youtao, Kato Akio, Jones Gary W.*. (-)-Epigallocatechin-3-gallate Inhibits Fibrillogenesis of Chicken Cystatin. J Agric Food Chem. 2015, 63(5): 1347-51.
- He Jianwei(*),Wang Yu,Chang Alan K.,Xu Linan,Wang Na,Chong Xiaoying,Li Hui,Zhang Bing,Jones GaryW.(*),Song Youtao(*),Myricetin Prevents Fibrillogenesis of Hen Egg White Lysozyme,J Agric Food Chem. 2014, 62(39): 9442-9449
- Chong Xiaoying, Lu Xian, Wang Yu, Chang Alan K., Xu Linan, Wang Nan, Sun Yonghui, Jones Gary W., Song Youtao, Song Yong-Bo, He Jianwei(*).Distinct structural changes in wild-type and amyloidogenic chicken cystatin caused by disruption of C95-C115 disulfide bond. J Biomol Struct Dyn. 2016, 5:1-9.
- He Jianwei,Xu Linan,Zou Zhiyuan,Ueyama N.,Li Hui,Kato Akio,Jones GW.,Song Youtao(*),Moleculardynamics simulation to investigate the impact of disulfide bond formation on conformational stability ofchicken cystatin I66Q mutant.,J Biomol Struct Dyn.,2013,31(10):1101-111
Federica Cossu
Institute of Biophysics at the National Research Council (IBF-CNR), Italy
Title: Type I BIR domain inhibitors in cancer therapy: designing drugs to modulate the NF-κB pathway
Time :

Biography:
Federica Cossu has always been interested in the field of cancer research, being fascinated by structural studies of crucial macromolecules and protein complexes involved in the cellular processes of cell death/survival. She gathered experience in cloning, expression, purification and crystallization of recombinant proteins, mainly belonging to the field of cancer. During the last years, she focused on the structure-based design of small molecules to be developed as drug candidates directed to pre-clinical studies. The success of this activity is proven by one patent, one award for her PhD thesis and several publications in the field. She progressively improved her knowledge on biophysical techniques for the study of proteins and on the in silico analysis of protein structures. She collected experiences in European laboratories, including several short visits/experiments at the ESRF synchrotron in Grenoble and at SOLEIL in Paris. She has been working in lab equipe for ten years, covering different positions within the research group, from master degree student to post-doc. She has been the supervisor of students, also giving lessons on the structural approaches applied to cancer therapy.
Abstract:
Inhibitors of apoptosis proteins (IAPs) constitute a family of conserved proteins whose over-expression enhances cell survival and resistance to anticancer agents. IAPs are E3 ligases, ubiquitylating substrates for the regulation of NF-kB; furthermore, they sequester caspases to prevent apoptosis. IAPs interactions occur through type I and type II BIR (Baculovirus IAP repeat) domains. Smac-mimetics (SM) mimicking the active N-terminal peptide of Smac-DIABLO, the natural antagonist of IAPs, have been shown to sensitize cancer cells to apoptosis. SM interact with type II BIR domains of IAPs, thus relieving caspases from X-linked IAP (XIAP) inhibitory activity and leading to cellular IAPs (cIAPs) auto-ubiquitylation and proteasomal degradation within minutes from exposure. Although SM are currently promising candidates for cancer therapy, some cancer cell lines present SM-resistance due to renewed cIAP2 activity and re-activation of NF-κB. IAPs-mediated regulation of NF-kB signaling is based on the formation of different protein-protein complexes, regulating ubiquitin-dependent signal transduction cascades. The type I BIR domain from different IAPs has been recognized as a pivotal platform for the assembly of such complexes.
We analyzed the surface of type I BIR domains (X- and cIAP-BIR1) to identify the hot-spots for the relevant protein-protein interactions. Virtual docking using libraries of compounds returned hits (NF023 and analogues) able to impair BIR1-based complexes with predicted low micromolar affinities that were experimentally confirmed. To this purpose, in vitro assays include fluorescence-based and biophysical techniques (Thermofluor, Microscale Thermophoresis, SEC, DLS, SLS). Crystallography on the protein-ligand complexes is the core of the structure-driven approach used for the iterative optimization of specific and selective drug candidates. Treatment of cancer cell cultures with the selected compounds will verify their effects on the modulation of IAPs-dependent signaling cascades. This represents a novel strategy to promote apoptosis in cancer and will unravel new insights on the regulation of NF-kB pathway.
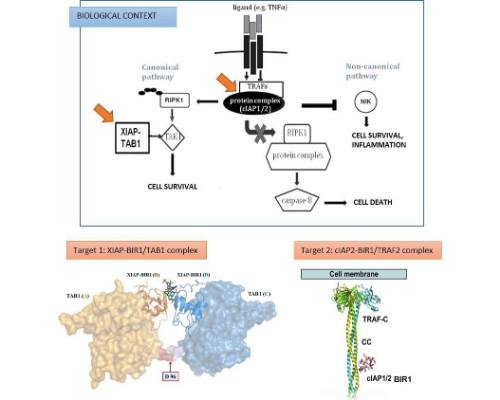
References:
- Federica Cossu, Milani M, Grassi S, Malvezzi F, Corti A, Bolognesi M and Mastrangelo E (2015) “NF023 binding to XIAP-BIR1: Searching drugs for regulation of the NF-κB pathway”. Proteins 83(4): 612-20
- Workshop by NanoTemper Technologies GmbH
Session Introduction
Tobias Pflüger
NanoTemper Technologies GmbH, Germany
Title: Solutions for studying protein complexes in structural biology and drug development
Biography:
Tobias Pflüger studied Chemistry at the Albert-Ludwigs-University in Freiburg with an emphasis on biochemistry. He received his PhD in Structural Biology investigating the structure and function of membrane proteins involved in cellular signalling cascades. In late 2015, Tobias joined NanoTemper Technologies as an application specialist.
Abstract:
Key to understanding the functional role of target proteins is elucidating the structure-function relationship between two proteins or protein complexes. Current analysis is often complex, time-consuming, and lacks data quality.
One of the goals of structural biology is to bring together multiple disciplines and methodologies as a means to better characterize proteins and protein complexes. For highly utilized techniques such as cryo-EM, X-ray crystallography and NMR, having the ability to identify and quantify binding affinities as well as analyze conformational and colloidal properties, enables researchers to gain a better understanding of the functional properties of protein targets.
During this workshop, you will learn about practical solutions to monitor and optimize protein stability and quality. An overview of the various analytical and biophysical methods commonly used by structural biologist will be discussed. We will share case studies demonstrating the benefits of understanding conformational and colloidal properties of protein complexes and how to use this information for further downstream analysis. Finally, we will share examples of protein-small molecule and protein-protein interactions and novel methodologies that assist researchers in making better decisions.
- Track 10: Structural Biology in Cancer Research | Track 11; Current Trends
Session Introduction
Guo-Ping Zhou
Gordon Life Science Institute, Boston, USA
Title: The NMR Studies of the Interaction between Polysialic Acid (polySA) and the PSTD Peptide in ST8Sia IV Polysialytranseferase

Biography:
Guo-Ping Zhou is currently a Distinguished Professor of Gordon Life Science Institute, USA. He is also an Adjunct Professor of several academics in the United States and China. Dr. Zhou received his Ph.D in Biophysics from University of California at Davis, and completed his postdoctoral training at Stanford University and Harvard University, respectively. Dr. Zhou determined the 3D NMR structures of some important proteins, protein-DNA complexes, and super lipids. He has successfully introduced the elegant wenxiang diagrams to elucidate the biological mechanisms of protein-protein/ligands interactions observed by NMR. Meanwhile, he has also published many papers in bioinformatics, and edited some special issues on structural biology for several influential scientific journals.
Abstract:
The α2,8-sialyltransferase (ST8Sia) family consists of 6 sia-lytranseferases, which are related to forms of polysialic acid chains (PSA) on neural cell adhesion molecule (NCAM) and NCAM polysialylation, and have important effects on formation of sialic acid storage diseases, neural system diseases and invasive cancers. It has been known that synthesis of PSA chains is catalysed by two polysialyltransferases, ST8Sia II (STX) and ST8Sia IV (PST). In addition, a polybasic motif of 32 amino acids in both ST8Sia II and ST8Sia IV has been designated as “polysialytransferase domain” (PSTD), which is essential for NCAM polysialylation. In this study, we have determined the 3D structure of the PSTD peptide containing 22 amino acids (22AA) in ST8Sia IV using NMR spectroscopy. This NMR-based model displays that the PSTD domain consists of an α-helical segment, two unstructured domains in both N- and C-terminus, and two three-residue-loops near the C-terminus of the peptide. Our overlaid 2D 1H-15N-HSQC spectra of the 22AA-PSTD peptide show that the amide proton chemical shifts of some amino acids such as I260, I261, H262, R265, L269 and K272 have been changed after polySA was mixed with the PSTD peptide. In addition, the peak intensity of A263, V264, R265, Y267, L269 and K272 were also decreased after adding polySA. However, there is no any change in both chemical shift and the amide proton peak intensity for all other residues located on outside of the helix. Above NMR results indicate a weak interaction exists between the helix of the PSTD and the PolySA, which may play a vital roles in modulating biosynthesis of polySA chain and NCAM polysialylation.
References:
- Huang RB, Cheng D, Lu B, Liao SM Troy li, FA, Zhou GP (2017) The Intrinsic Relationship between Structure and Function of the Sialytransferase ST8Sia Family Members. Curr. Top. Med. Chem. 7(21):2359-2369.
- Zhou G P, W Z Zhong (2016). Perspectives in the medicinal chemistry. Current Topics in Medicinal Chemistry.16(4):381-382.
- Zhou G P (2016) Modulations and their biological functions of protein-biomolecule interactions. Current Topics in Medicinal Chemistry. 16(6):579-580.
- Zhou G P, Chen D, Liao S, Huang R B (2016) Recent progresses in studying helix-helix interactions in proteins by incorporating the Wenxiang diagram into the NMR spectroscopy. Current Topics in Medicinal Chemistry, 16(6)581-590.
- Zhou G P, Huang R B, Troy, F A (2015) 3D Structural Con formation and Functional Domains of Polysialyltransferase ST8Sia IV Required for Polysialylation of Neural Cell Adhesion Molecules. Protein Peptide Lett. 22(2):137-148.
Ozge Sensoy
Istanbul Medipol University, Turkey
Title: Understanding Differential Selectivity of Arrestins toward the Phosphorylation State of G-protein-coupled Receptors

Biography:
As being a computational biophysicist, my research has focused on understanding underlying molecular mechanisms of biologically important problems and also providing mechanistic insight at the molecular level. In particular, I have been working with GPCRs and their interacting partners which are responsible for cellular signaling. In order to complement relevant experimental studies one needs to access long time-scales and big system sizes which are beyond the classic MD simulations. In this respect, my expertise in doing long-time MD simulations and application of enhanced sampling techniques such as accelerated MD, metadynamics, and steered MD which provides a good fit. I work in close collaboration with medicinal chemists to direct them for effective molecular designs. In addition, I am also responsible for testing the efficacy of these molecules in silico before transferring them to either in vitro or in vivo studies. Recently, I have been awarded an international COST (European Cooperation in Science and Technology) grant which is based on developing heterobivalent molecules capable of binding more than one target for treatment of symptoms of Parkinson’s disease.
Abstract:
Arrestins (Arrs) are a family of four proteins (Arr1- 4) which mediate G-protein-coupled receptor (GPCR) desensitization and internalization by coupling to active and phosphorylated receptor. Recently, they have also been shown to mediate GPCR-independent signaling pathways. The specific functions of Arrs (desensitization vs. G-protein-independent signaling) can be regulated by differential phosphorylation of the receptor, which is known as the phosphorylation barcode. The molecular mechanism responsible for formation of a high-affinity complex between an Arr subtype and a GPCR having a certain phosphorylation pattern remains elusive but is crucial for directing the subtype towards a specific functional role, and hence paves the way for development of safer therapeutics with fewer side-effects. As a first step in that direction, we have started with elucidating the activation mechanism of Arr subtypes by carrying out comparative molecular dynamics (MD) studies of the two members of the family, namely Arr1 and Arr3, which exhibit the largest differences in terms of phosphorylation selectivity. In addition, we also modeled and simulated Arr1-R175E mutant, which is known to be constitutively active, and compared it to Arr1 and Arr3 to detect activation-related rearrangements. We found novel structural elements that had not been considered before as determinants for activation and can be targeted with drugs for functional modulation. The emerging model also proposes that activation of Arr1-R175E is connected to perturbation of the well-known region, namely, the polar-core, whereas no changes were observed in that region in Arr3 in spite of the presence of other activation-related changes. With that, we could propose a structural model to explain the molecular mechanism responsible for markedly reduced selectivity of Arr3 towards phosphorylated GPCRs. Finally, knowledge achieved in this study can also be utilized to modulate Arr binding to GPCRs under disease conditions such as otozomal dominant disorders and congestive heart failure.
References:
- Sensoy O, Atılgan A R and Atılgan C (2017) FbpA iron storage and release are governed by periplasmic microenvironments. PCCP. 19(8): 6064-6075.
- Sensoy O, Moreira I S and Morra G (2016) Understanding the Differential Selectivity of Arrestins toward the Phosphorylation State of the Receptor. ACS Chemical Neuroscience. 7(9):1212.
- Arango Lievano M, Sensoy O, Borie A, Corbani M, Guillon G, Sokoloff P and Weinstein H, Jeanneteau F (2015) A GIPC1-Palmitate Switch Modulates Dopamine DRD3 Receptor Trafficking and Signaling. Mol. Cell. Biol. 36(6):1019-1031.
- Sensoy O and Weinstein H (2015) A mechanistic role of Helix 8 in GPCRs: Computational modeling of the Dopamine D2 Receptor interaction with the GIPC1-PDZ domain. BBA-Biomembranes. 1848(4):976-983.
- Dalgicdir C Sensoy O and Sayar M (2013) A transferable coarse-grained model for diphenylalanine: how to represent an environment driven conformational transition. J. Chem. Phys. 139(23):234115.
Stephen M. Soisson
Merck Research Laboratories, USA
Title: Structural Basis for the Cooperative Allosteric Activation of the Free Fatty Acid Receptor GPR40
Biography:
Stephen Soisson, PhD is a director of Biochemical Engineering and Structure at Merck Research Laboratories in West Point, Pennsylvania (USA). With 25+ years of structural biology experience, Dr. Soisson has focused research on elucidating the structural aspects of biological regulatory mechanisms, and applying these insights in the area of structure-based drug design. He has served on the scientific advisory boards of the Structural Genomics Consortium, and the GPCR Consortium.
Abstract:
Clinical studies indicate that partial agonists of the G-protein-coupled, free fatty acid receptor GPR40 enhance glucose-dependent insulin secretion and represent a potential mechanism for the treatment of type 2 diabetes mellitus. Recently identified, full allosteric agonists (AgoPAMs) of GPR40 bind to a site distinct from partial agonists and can provide additional efficacy. Our recent studies have led to a 3.2-Å crystal structure of human GPR40 (hGPR40) in complex with both the partial agonist MK-8666 and an AgoPAM. Surprisingly, the structure reveals a novel lipid-facing AgoPAM-binding pocket outside the transmembrane helical bundle. Comparison with an additional 2.2-Å structure of the hGPR40–MK-8666 binary complex reveals an induced-fit conformational coupling between the partial agonist and AgoPAM binding sites, involving rearrangements of the transmembrane helices 4 and 5 (TM4 and TM5). These structural rearrangements, along with AgoPAM binding, appear to trigger the transition of intracellular loop 2 (ICL2) into a short helix. These conformational changes likely prime GPR40 to a more active-like state and explain the binding cooperativity between these ligands.
References:
- NL Elsen, SB Patel, RE Ford, DL Hall, F Hess, H Kandula, M Kornienko et. al. (2017) Insights into activity and inhibition from the crystal structure of human O-GlcNAcase. Nature Chemical Biology. 13 (6):613-615.
- H Zhang, GW Han, A Batyuk, A Ishchenko, KL White, N Patel et. al. (2017) Structural basis for selectivity and diversity in angiotensin II receptors. Nature. 544(7650):327-332.
- HP Su K Rickert, C Burlein, K Narayan, M Bukhtiyarova, DM Hurzy, et. al. (2017) Structural characterization of nonactive site, TrkA-selective kinase inhibitors. Proceedings of the National Academy of Sciences. 114(3):297-306.
- M Scheepstra, S Leysen, GC Van Almen, JR Miller, J Piesvaux, V Kutilek, et. al. (2015) Identification of an allosteric binding site for RORγt inhibition. Nature communications. 6:8833.
Biography:
Abstract:
Andrei Korostelev
UMass Medical School, USA
Title: Conformational dynamics revealed by ensemble cryo-EM

Biography:
Andrei A. Korostelev is passionate about mechanisms of translation regulation. He received Ph.D. in Michael S. Chapman laboratory at Florida State University in 2003 and performed postdoctoral studies with Harry F. Noller in 2004-2010. The Korostelev laboratory at the RNA Therapeutics Institute uses recent advances in biochemical and structural methods to elucidate detailed mechanisms that govern translation and regulation of translation under stress conditions or during disease. Recent work revealed high-resolution “frames” of the motions that the translational machinery undergoes during bacterial stress responses (including the stringent response) and viral infection, as summarized on the laboratory web site: http://labs.umassmed.edu/korostelevlab/research.htm
Abstract:
Virus propagation depends on efficient synthesis of viral proteins by the host translational machinery. Internal ribosome entry sites (IRESs) of viral mRNAs mediate cap-independent initiation. Intergenic-region (IGR) IRESs of Dicistroviridae family, which includes the Taura syndrome virus (TSV) and Cricket paralysis virus (CrPV), use the most streamlined mechanism of initiation, independent of initiation factors and initiator tRNA. A tRNA-mRNA like pseudoknot of IGR IRESs binds the ribosomal A (aminoacyl-tRNA) site of the 80S ribosome (Fernandez et al., 2014; Koh et al., 2014). The pseudoknot has translocate to the P site to allow binding of the first tRNA and initiate translation.
Using electron cryo-microscopy of a single specimen, we resolved five ribosome structures formed with the Taura syndrome virus IRES and translocase eEF2•GTP bound with sordarin. The structures suggest a trajectory of IRES translocation, required for translation initiation, and provide an unprecedented view of eEF2 dynamics (animation).
The IRES rearranges from extended to bent to extended conformations. This inchworm-like movement is coupled with ribosomal inter-subunit rotation and 40S head swivel. eEF2, attached to the 60S subunit, slides along the rotating 40S subunit to enter the A site. Its diphthamide-bearing tip at domain IV separates the tRNA-mRNA-like pseudoknot I (PKI) of the IRES from the decoding center. This unlocks 40S domains, facilitating head swivel and biasing IRES translocation via hitherto-elusive intermediates with PKI captured between the A and P sites.
References:
- Demo G, Svidritskiy E, Madireddy R, Diaz-Avalos R, Grant T, Grigorieff N, Sousa D, Korostelev AA. Mechanism of ribosome rescue by ArfA and RF2. preprint in bioRxiv. 2016 Dec. 2. (animation)
- Loveland AB, Bah E, Madireddy R, Zhang Y, Brilot AF, Grigorieff N, Korostelev AA. Ribosome•RelA structures reveal the mechanism of stringent response activation. eLife. 2016 July 19. (animation) animation)
- Abeyrathne PD, Koh CS, Grant T, Grigorieff N, Korostelev AA. Ensemble cryo-EM uncovers inchworm-like translocation of a viral IRES through the ribosome. eLife. 2016 May 9. (animation)
- Svidritskiy E, Madireddy R, Korostelev AA. Structural Basis for Translation Termination on a Pseudouridylated Stop Codon. J Mol Biol. 2016 Apr 20.
- Svidritskiy E, Korostelev AA. Ribosome Structure Reveals Preservation of Active Sites in the Presence of a P-Site Wobble Mismatch. Structure. 2015 Nov 3;23(11):2155-61.
Tzu-Ching Meng
Academia Sinica, Taiwan
Title: Structural basis for PTPN3-p38gamma complex involved in colon cancer progression

Biography:
Tzu-Ching has completed his PhD from University of Nebraska Medical Center in 1999 and postdoctoral studies from Cold Spring Harbor Laboratory in 2003. Since then, he has been working at Academia Sinica, the premier government-funded institution in Taiwan. He is now a Research Fellow with professorship jointly appointed by National Taiwan University. He has published more than 40 papers in reputed journals and has been serving as an advisory board member of competitive journals.
Abstract:
The Ras signaling cascade acts as a key driver in human colon cancer progression. Among the modules in this pathway, p38gamma (MAPK12) and its specific protein tyrosine phosphatase PTPN3 (PTPH1) are critical regulators responsible for Ras oncogenic activity. However, the molecular basis for their interaction is completely unknown. Here we report the unique architecture of the PTPN3-p38gamma complex by employing an advanced hybrid method integrating X-ray crystallography, small-angle X-ray scattering (SAXS) and chemical cross-linking/mass spectrometry (CX-MS). Our crystal structure of PTPN3 in complex with the p38gamma phosphopeptide presented a unique feature of the E-loop that defines the substrate specificity of PTPN3 towards fully activated p38gamma. The low-resolution structure demonstrated the formation of an active-state or a resting-state complex of PTPN3-p38gamma. We showed a regulatory function of PTPN3’s PDZ domain, which stabilizes the active-state complex through interaction with the PDZ-binding motif of p38gamma. Using SAXS and CX-MS approaches, we found that binding of the PDZ domain to the PDZ-binding motif lifts the atypical auto-inhibitory constraint of PTPN3, enabling efficient tyrosine dephosphorylation of p38gamma to occur. Our findings emphasize the potential of structural approach for PTPN3-p38gamma complex that may deliver new therapeutic strategies against Ras-mediated oncogenesis in colon cancer.
References:
- Pan KT, Chen YY, Pu TH, Chao YS, Yang CY, Bomgarden RD, Rogers JC, Meng, TC*, Khoo, KH* (2014) Mass spectrometry based quantitative proteomics for dissecting multiplexed redox cysteine modifications in nitric oxide-protected cardiomyocyte under hypoxia. Antioxidant and Redox Signaling, 20:1365-1381.
- Santhanam A, Peng WH, Yu YT, Sang TK, Chen GC*, Meng TC* (2014) Ecdysone-induced receptor tyrosine phosphatase PTP52F regulates Drosophila midgut histolysis by enhancement of autophagy and apoptosis. Mol. Cell. Biol., 34:1594-1606.
- Chen KE, Lin SY, Wu M J, Ho MR, Santhanam A, Chou CC, Meng TC*, Wang AHJ* (2014) Reciprocal allosteric regulation of p38γ and PTPN3 involves a PDZ domain modulated complex formation. Science Signaling, 7: ra98 p1-12.
- Chen KE, Li MY, Chou CC, Ho MR, Chen GC, Meng TC*, Wang AHJ* (2015) Substrate specificity and plasticity of FERM-containing protein tyrosine phosphatases. Structure, 23: 653–664.
- Hsu MF, Pan KT, Chang FY, Khoo KH, Urlaub H, Chang GD*, Haj FG*, and Meng TC* (2016) S-Nitrosylation of endogenous protein tyrosine phosphatases in endothelial insulin signaling. Free Rad Biol Med, 99: 199-213.
Christian Biertümpfel
Max Planck Institute of Biochemistry, Germany
Title: Holliday junction resolvase GEN1 functions as a versatile DNA sensor and processor

Biography:
Christian Biertümpfel obtained his Ph.D. degree from the European Molecular Biology Laboratory (EMBL) and the Ruprecht Karls University of Heidelberg, Germany. His Ph.D. research focused on the crystallization and characterization of Holliday junction resolvases. During his postdoctoral time at the National Institute of Diabetes and Digestive and Kidney Diseases, NIH, Bethesda, MD, USA, he was able to solve a first crystal structure of a Holliday junction resolvase from T4 phages in complex with a DNA four-way junction. Furthermore, together with Wei Yang he determined the structure and mechanism of human DNA polymerase η functioning as a molecular splint. After a short period at the Vaccine Research Center, NIAID, NIH, he moved to the Max Planck Institute of Biochemistry, Martinsried, Germany as a Max Planck Research Group Leader. Recently, the Biertümpfel lab obtained structural information on the human Holliday junction resolvase GEN1 and they found for the first time a chromodomain extending a nuclease domain.
Abstract:
Several DNA repair and maintenance pathways depend on the correct and efficient processing of DNA intermediates by structure-specific nucleases. Human Holliday junction resolvase GEN1 seems to be an enzyme of last resort for recognizing and cleaving a specific range of DNA structures. The crystal structure of human GEN1 in complex with Holliday junction DNA pinpointed to a crucial role of the chromodomain for efficient DNA recognition and cleavage. We further characterized different DNA-binding modes of GEN1 using biochemical methods in combination with structure-guided mutagenesis. The analysis highlights the importance of the arch region to distinguish between different DNA substrates. In addition, we identified a cluster of positive amino acids shadowing the chromodomain to assist the enzyme for robust DNA recognition. Moreover, we directly show that GEN1 operates as a monomer with 5’ flap DNA and as a dimer in complex with DNA four-way junctions, which is a unique feature in the Rad2/XPG nuclease family. This linked cleavage mechanism ensures that DNA junctions are resolved in a strictly symmetric manner without altering DNA information. GEN1’s DNA recognition features make it a versatile tool for DNA processing and for maintaining genome integrity.

Figure: Holliday junction resolvase GEN1 is a monomer in solution and thus, cleavage competent for 5’ flap substrates. However, it can only cleave DNA four-way junctions by forming an active nuclease dimer.
References:
- Lee SH, Princz LN, Klügel MF, Habermann B, Pfander B, Biertümpfel C. (2015). Human Holliday junction resolvase GEN1 uses a chromodomain for efficient DNA recognition and cleavage. Elife, 4, e12256.
- Zhao Y, Gregory MT, Biertümpfel C, Hua YJ, Hanaoka F, Yang W. (2013). Mechanism of somatic hypermutation at the WA motif by human DNA polymerase η. Proc Natl Acad Sci U S A 110, 8146-51.
- Joyce MG, Kanekiyo M, Xu L, Biertümpfel C, Boyington JC, Moquin S, Shi W, Wu X, Yang Y, Yang ZY, Zhang B, Zheng A, Zhou T, Zhu J, Mascola JR, Kwong PD, Nabel GJ. (2013). Outer domain of HIV-1 gp120: antigenic optimization, structural malleability, and crystal structure with antibody VRC-PG04. J Virol 87, 2294-306.
- Zhao Y, Biertümpfel C, Gregory MT, Hua YJ, Hanaoka F, Yang W. (2012). Structural basis of human DNA polymerase η-mediated chemoresistance to cisplatin. Proc Natl Acad Sci U S A 109, 7269-74.
- Hansman GS, Biertümpfel C, Georgiev I, McLellan JS, Chen L, Zhou T, Katayama K, Kwong PD. (2011). Crystal structures of GII.10 and GII.12 norovirus protruding domains in complex with histo-blood group antigens reveal details for a potential site of vulnerability. J Virol 85, 6687-701
Paola Picotti
ETH Zurich, Switzerland
Title: Monitoring protein structural changes on a proteome-wide scale
Biography:
After her PhD at the University of Padua (Italy) Paola Picotti joined the group of Ruedi Aebersold at ETH Zuerich (Switzerland), where she developed novel targeted proteomic techniques. In 2011 she was appointed assistant professor at ETH Zurich. Her group develops structural and chemoproteomics methods and uses them to study the consequences of intracellular protein aggregation. Paola Picotti’s research was awarded an ERC Starting grant, a Professorship grant from the Swiss National Science Foundation, the Latsis Prize, the Robert J. Cotter Award, the SGMS Award and the EMBO Young Investigator Award. Main contributions of the Picotti group are the development of a structural method to analyze protein conformational changes on a system-wide level, the discovery of novel allosteric interactions, the analysis of the determinants of proteome thermostability and the identification of a novel neuronal clearance mechanism for a protein involved in Parkinson’s disease.
Abstract:
Protein structural changes induced by external perturbations or internal cues can profoundly influence protein activity and thus modulate cellular physiology. Mass spectrometry (MS)-based proteomic techniques are routinely used to measure changes in protein abundance, post-translational modification and protein interactors, but much less is known about protein structural changes, owing to the lack of suitable approaches to study global changes in protein folds in cells.
In my talk I will present a novel structural proteomics technology developed by our group that enables the analysis of protein structural changes on a proteome-wide scale and directly in complex biological extracts. The approach relies on the coupling of limited proteolysis (LiP) tools and an advanced MS workflow. LiP-MS can detect subtle alterations in secondary structure content, larger scale movements such as domain motions, and more pronounced transitions such as the switch between folded and unfolded states or multimerization events. The method can also be used to pinpoint protein regions undergoing a structural transition with peptide-level resolution. I will describe selected applications of the approach, including 1. The identification of proteins that undergo structural rearrangements in cells due to a nutrient shift; 2. The analysis of in vivo protein aggregation; 3. The cell-wide analysis of protein thermal unfolding; and 4. The identification of protein-small molecule interactions (e.g drug-target deconvolution).
I will discuss the power and limitations of the method and possible new directions in structural biology enabled by this emerging approach to protein structure analysis.
References:
- Leuenberger P, Ganscha S, Kahraman A, Cappelletti V, Boersema P,J, von Mering C, Claassen M, Picotti P. Cell-wide analysis of protein thermal stability across species reveals the determinants of thermostability, Science, (in press).
- Feng Y, De Franceschi G, Kahraman A, Soste M, Melnik A, Boersema PJ, de Laureto PP, Nikolaev Y, Oliveira AP, Picotti P. Global analysis of protein structural changes in complex proteomes. Nat Biotechnol. 2014; 32(10):1036-44.
- Soste M, Hrabakova R, Wanka S, Melnik A, Boersema P, Maiolica A, Wernas T, Tognetti M, von Mering C, Picotti P. A sentinel protein assay for simultaneously quantifying cellular processes. Nat Methods. 2014; 11(10):1045-8.
- Picotti P, Clément-Ziza M, Lam H, Campbell DS, Schmidt A, Deutsch EW, Röst H, Sun Z, Rinner O, Reiter L, Shen Q, Michaelson JJ, Frei A, Alberti S, Kusebauch U, Wollscheid B, Moritz RL, Beyer A, Aebersold R. A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis. Nature. 2013; 14;494(7436):266-70.
- Picotti P, Bodenmiller B, Mueller LN, Domon B, Aebersold R. Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell. 2009; 21;138(4):795-806.
Hagen Hofmann
Weizmann Institute of Science, Israel
Title: Slow domain reconfiguration causes power law kinetics in a two-state enzyme

Biography:
Hagen Hofmann received his PhD from the Martin-Luther University Halle-Wittenberg (Germany) in 2008. In the period 2008 - 2014, he was a postdoctoral fellow at the University of Zurich in the group of Benjamin Schuler and since 2014 he is heading the “Molecular Systems Biophysics” group at the Weizmann Institute of Science (Israel). He and his group use a broad set of single-molecule fluorescence tools to understand the dynamics of proteins and protein networks on timescales from nanoseconds to hours. In addition, live-cell imaging, in vivo single-molecule FRET, and single particle tracking is used to monitor proteins in live cells. His interest ranges from the physics of disordered proteins over coupled binding and folding reactions up to stochastic genetic circuits and regulatory protein networks.
Abstract:
Conformational transitions in proteins are typically captured well by rate equations that predict exponential kinetics for two-state reactions. Here, we describe a remarkable exception. The electron-transfer enzyme quiescin sulfhydryl oxidase (QSOX), a natural fusion of two functionally distinct domains, switches between open and closed domain arrangements with apparent power law kinetics. Using single-molecule Foerster resonance energy transfer (FRET) experiments on timescales from nanoseconds to milliseconds, we show that the unusual open-close kinetics results from slow domain rearrangements in a heterogeneous ensemble of open conformers. While substrate accelerates the kinetics, thus suggesting a substrate-induced switch to an alternative free energy landscape of the enzyme, the power-law behavior is also preserved upon electron load. Our results show that conformational multiplicity with slow sampling dominates the motions of QSOX, thus providing an explanation for catalytic memory effects in other enzymes.
References:
- Grossman, I. et al. (2015) Single-molecule spectroscopy exposes hidden states in an enzymatic electron relay. Nature Communications 6:1-10.
- Hofmann, H (2016). Speedy Motion For Function. Nature Chemical Biology. 12:576-577.
- Schuler, B; Soranno, A; Hofmann, H; Nettels, D (2016). Single-Molecule Fret Spectroscopy and the Polymer Physics of Unfolded and Intrinsically Disordered Proteins. Annual Review of Biophysics. 45:207-231.
- Hofmann, H; Soranno, A; Borgia, A; Gast, K; Nettels, D; Schuler, B (2012). Polymer Scaling Laws of Unfolded and Intrinsically Disordered Proteins Quantified With Single-Molecule Spectroscopy. Proceedings of the National Academy of Sciences of the United States of America. 109:16155-16160.
5. Hofmann, H; Hillger, F; Pfeil, Sh; Hoffmann, A; Streich, D; Haenni, D; Nettels, D; Lipman, Ea; Schuler, B (2010). Single-Molecule Spectroscopy of Protein Folding in a Chaperonin Cage. Proceedings of the National Academy of Sciences of the United States of America. 107:11793-11798.
Marie Chabbert
University of Angers, France
Title: Functional divergence in protein families: A co-variation analysis
Biography:
Marie Chabbert is a scientist from the French CNRS (Centre National de la Recherche Scientifique). She has her expertise in molecular modeling and bioinformatics approaches to the structure-function relationship of proteins. She has special interest in deciphering the mechanisms that drove protein evolution and in using evolutionary data to gain structural and functional information on protein families. She is presently working on the G protein-coupled receptors, especially chemotaxic and vasoactive peptide receptors.
Abstract:
Statement of the Problem: Co-variations between positions in a multiple sequence alignment may reflect structural, functional, and/or phylogenetic constraints. Numerous co-variation methods have been developed and may yield a wide variety of results. However, few studies have been undertaken to determine co-variations methods adequate to gain information on functional divergence within a protein family. Methodology & Theoretical Orientation: We explore co- variation methods for their capability to mine co-varying positions related to the functional divergence in a protein family. To reach this objective, we compare several methods on a model system that consists of three nested sets of about 300, 100, and 20 paralogous sequences of a protein family, the class A of G protein-coupled receptors. The co- variation methods analyzed are based on chi2 scores, mutual information, substitution matrices, or perturbation methods. We analyze the dependence of the co-variation scores on residue conservation, measured by sequence entropy, and the networking structure of the top pairs. Findings: Out of the four methods that privilege top pairs with intermediate entropy, two favor individual pairs, whereas the other two methods, OMES (Observed minus Expected Squared) and ELSC (Explicit Likelihood of Subset Covariation), favor a network structure with a central residue involved in several high scoring pairs. This network structure is observed for the three sequence sets, making a hierarchical analysis possible. In each case, the central residue corresponds to a residue known to be crucial for the evolution of the protein family and the sub-family specificity. Positions co-varying with this central residue form a few clusters in the receptor 3D structure (Fig. 1). Conclusion & Significance: The central residues obtained with the OMES or ELSC methods can be viewed as evolutionary hubs, in relation with an epistasis-based mechanism of functional divergence within a protein family.
References:
- Chantreau V., Taddese B., Munier M., Gourdin L., Henrion D., Rodien P. and Chabbert M. (2015) Molecular Insights into the Transmembrane Domain of the Thyrotropin Receptor, Plos One, 10(11):e0142250.
- Pelé J., Moreau M., Abdi H., Rodien P., Castel H. and Chabbert M. (2014) Comparative analysis of sequence co-variation methods to mine evolutionary hubs: Examples from selected GPCR families. PROTEINS 82:2141-56.2013.
- Pelé J., Bécu J.-M., Abdi H. and Chabbert M. (2012) Bios2mds: an R package for comparing orthologous protein families by metric multidimensional scalingBMC Bioinformatics 13, 133.
- Chabbert M., Castel H., Pelé J., Devillé J., Legendre R. and Rodien P. (2012) Evolution of Class A G-Protein-Coupled Receptors: Implications for Molecular Modeling. Curr. Med. Chem. 19, 1110-8.
- Pelé J., Abdi H., Moreau M., Thybert D., and Chabbert M. (2011) Multidimensional scaling reveals the main evolutionary pathways of class A G-protein-coupled receptors. (2011) PLoS ONE 6, e19094.
CongBao Kang
Agency for Science, Technology and Research (A*STAR), Singapore
Title: Structural and dynamic studies of DENV and ZIKV proteases and its insight into inhibitor design

Biography:
CongBao Kang received his Ph.D. from School of Biological Sciences at Nanyang Technological University (NTU). He was a research fellow at Centre for Structural Biology, Vanderbilt University, where he was working on structural determination of disease-related membrane proteins. He is currently the group leader of high End NMR group at ETC. His group is working on protein structure, dynamics and its interaction with potential drug candidates using solution NMR spectroscopy. The goal of his group is to provide structural information of a target protein to the medicine chemists to understand structure-activity relationship of potent compounds. His group is involving in hits identification, hits to lead, and lead optimization steps of the drug discovery process. His is currently working on target-based drug discoveries. The targets include methyltransferases, kinases, ion channels, membrane-bound receptors, protein-protein interactions, and viral proteins.
Abstract:
Dengue virus (DENV and Zika virus (ZIKV) belong to Flaviviridae genus which contains important human pathogens. DENV affects people living in tropical and subtropical regions. DENV infection can cause serious diseases such as dengue fever. ZIKV has drawn worldwide attention because of the outbreak in 2015. Viral genome of a flavivirus encodes a polyprotein that can be processed into structural and non-structural (NS) proteins by both host and viral proteases. Viral protease is a two-component serine protease formed by a cofactor region (~40 aa) from NS2B and a protease region (~170 aa) from NS3. The NS2B-NS3 protease of DENV or ZIKV is a validated target because of their function in maturation of viral proteins. Structural studies have been conducted for both DENV and ZIKV proteases. For DENV, previous studies have demonstrated that the free protease adopts an open conformation in which the C-terminal part of the NS2B cofactor region stays away from the active site. In the presence of an inhibitor, DENV protease forms a closed conformation in which the C-terminal region of NS2B forms part of the active site and interacts with the inhibitor. Our NMR study reveals that an unlinked DENV protease adopts the closed conformation in solution. Based on the knowledge on DENV protease, several constructs were made for ZIKV protease. Structural studies demonstrated that ZIKV protease adopts the closed conformation in the absence and presence of an inhibitor or substrate. The linker or enzymatic cleavage site present between NS2B and NS3 may affect inhibitor to interact with the active site. Our accumulated studies have shown that the unlinked protease construct can be used for studying protease-inhibitor interactions. We have demonstrated that the unlinked ZIKV protease interacts with different types of inhibitors. Our studies will be helpful for structure-based inhibitor design against both ZIKV and DENV proteases.
References:
- Zhang Z, Li Y, Loh YR, Phoo WW, Hung AW, Kang C*, Luo D* (2016) Crystal structure of unlinked NS2B-NS3 protease from Zika virus. Science, 354(6319):1597-1600.
- W. W. Phoo, Y. Li, Z. Z. Zhang, M. Y. Lee, Y. Loh, Y. B. Tan, E. Y. Ng, J. Lescar, C. Kang*, D. Luo* (2016) Structure of NS2B-NS3 Protease from Zika Virus after Self-cleavage. Nature communications, 7:13410.
- Y. Li, YL Wong, M.Y. Lee, Q. Li, J. Lescar, P.Y. Shi, C. Kang*, (2016) Secondary structure and membrane topology of the full length Dengue NS4B in micelles. Angewandte Chemie Int Ed Engl, 55(39):12068-72
- Y. Li, Q. Li, Y.L. Wong, L. Liew, and C.B. Kang,*(2015) Membrane topology of NS2B of dengue virus revealed by NMR spectroscopy. BBA-Biomembranes. 1848: 2244-2252.
- Zou J, Xie X., Lee LT., Chandrasekaran R., Reynaud A., Yap L., Wang Q., Dong H., Kang C.B., Yuan Z., Lescar J., and Shi P. (2014) Dimerization of Flavivirus NS4B Protein. Journal of Virology. 88(6): 3379-3391.
Jungsan Sohn
Johns Hopkins University, USA
Title: Assembly mechanism of foreign dsDNA-sensing inflammasomes
Biography:
The research focus in the Sohn is to understand the molecular mechanism by which human immune system engages invading pathogens. Jay received B.S. from University of Michigan, and Ph.D. from Duke University. Upon completing his post-doctoral training at MIT with Dr. Bob Sauer, he joined the faculty at Johns Hopkins in 2011.
Abstract:
Absent-in-melanoma-2-like receptors (ALRs) detect foreign double-stranded (ds)DNA from invading pathogens and assemble into filamentous signaling platforms termed inflammasome. The ALR filaments play crucial roles in launching antiviral and inflammatory responses against a number of pathogens (e.g. HIV and HSV); however, persistent ALR complexes are also linked to autoimmune disorders (e.g. Sjögren’s syndrome and lupus). Here, by combining solution assays, electron microscopy, and single-molecule methods, we investigate the filament assembly mechanisms of two prototypical ALRs, namely IFI16 and AIM2.
(1) IFI16 detects foreign dsDNA both in the host nucleus and cytoplasm. We found that IFI16 uses dsDNA as a one-dimensional diffusion-scaffold to assemble into filaments. The dsDNA-binding HIN200 domains of IFI16 are responsible for tracking dsDNA, while its pyrin domain (PYD) is necessary for filament assembly. Importantly, nucleosomes represent barriers that prevent IFI16 from targeting host dsDNA by directly interfering with its assembly. This unique scanning-assisted assembly mechanism would allow IFI16 to distinguish self- from nonself-dsDNA in the nucleus.
(2) AIM2 detects cytoplasmic dsDNA and assembles into an inflammasome. We found that the PYD of AIM2 (AIM2PYD) drives both filament formation and dsDNA binding. As with IFI16, the size of exposed dsDNA acts a key regulator for the polymerization of AIM2. The helical symmetry of the upstream AIM2PYD filament is consistent with the filament assembled by the PYD of the downstream ASC adaptor, indicating that AIM2 acts as a structural template for polymerizing ASC.
Together, our studies provide a unifying paradigm for how ALRs carry out foreign dsDNA-sensing pathways, where generating a structural template by coupling ligand-binding and oligomerization plays a key signal transduction mechanism.

References:
- Morrone, S.M., Wang, T., Constantoulakis, L.M., Hooy, R.M., Delannoy, M.R., and Sohn, J. (2014) Cooperative assembly of IFI16 filaments on dsDNA provides insights into host defense strategy. PNAS 111, E62-71
- Geertsma, H.J., Schute, A.C., Spenkelink, L.M., Mcgrath, W.J., Morrone, S.R., Sohn, J., Mangel, W.F., Robinson, A., van Oijen, (2015) A. Single-molecule imaging at high fluorophore concentrations of Dye (LaDYe). Biophysical Journal 108, 949-56.
- Baer, A.N., Petri, M.A., Sohn, J., Rosen, A., Casciola-Rosen, L. Antibodies to human IFI16 are present in systemic lupus and primary Sjögren’s syndrome with similar frequencies but detect different parts of molecule. (2015) Arthritis Care Res.
- Morrone, S.M. Matyszewski, M., Yu, X., Delannoy, M., Egelman, E.H., and Sohn J. Assembly driven activation of the AIM2 inflammasome provides a template for the polymerization of downstream ASC. (2015) Nature Communications. 6, 7827.
- Stratmann, S., Morrone, S.M., van Oijen, A.M.*, and Sohn, J*. The innate immune sensor IFI16 recognizes foreign DNA in the nucleus by scanning along the duplex. (2015). eLife e11721 *: co-corresponding authors
Francesca Massi
University of Massachusetts Medical School, USA
Title: RNA-binding domain disorder modulates the RNA destabilizing activity in the TTP family of proteins
Biography:
Abstract:
Despite the importance of RNA-binding proteins to gene regulation, our understanding of how their structure and dynamics contribute to their biological activity is limited. In this study, we focus on two related RNA-binding proteins—TTP and TIS11d—that regulate the stability of mRNA transcripts encoding key cancer-related proteins, such as tumor necrosis factor-a and vascular endothelial growth factor. These two proteins display differential folding propensity in the absence of RNA, despite sharing a high sequence identity. We identified three residues located at the C-terminal end of an a-helix that determine the folding propensity of the RNA-binding domain in the apo state. We also showed that stabilization of the structure of the RNA-binding domain is associated with differences in RNA-binding activity in vitro and increased RNA-destabilizing activity in the cell. Phylogenetic analysis indicates that this family of proteins has only recently evolved to be able to modulate its biological activity through its dynamic structure.
To investigate how three residues determine the folding and stability of the TZF domain we used molecular dynamics and NMR spectroscopy. We observed that a p-p stacking between the side chains of a conserved phenylalanine and the zinc coordinating histidine is essential to maintain the correct tetrahedral geometry between the three cysteines, the histidine and the zinc ion. A hydrogen bond in the C-terminal zinc finger of TIS11d is important to keep the phenylalanine in proximity of the imidazole ring of the zinc coordinating histidine in a conformation that allows for stacking of the side chains. Lack of this hydrogen bond in TTP is responsible for the reduced zinc affinity of the C-terminal zinc finger. Sequence alignment shows that this phenylalanine residue is highly conserved. These results suggest that most CCCH-type zinc finger proteins employ p-p interactions to stabilize the structure of the TZF domain.

Figure 1: The stacking of the aromatic rings of the conserved Phe and of the zinc coordinating His stabilizes conformation of the His in a rotameric state compatible for zinc-binding.
References:
- Morgan, B. R.; Massi, F. (2010) A computational study of RNA binding specificity in the tandem zinc finger domain of TIS11d. Protein Sci. 19: 1222-1234
- Morgan, B. R.; Massi, F. (2010) Accurate estimates of free energy changes in charge mutations. J. Chem. Theory Comput. 6: 1884-1893.
- Morgan, B. R.; Deveau, L. M.; Massi, F. (2015) Probing the structural and dynamical effects of the charged residues of the TZF domain of TIS11d. Biophys. J. 108: 1503-1515.
- Tavella, D.; Deveau, L. M.; Massi, F. (2016) Understanding the origin of the disorder of the tandem zinc finger domain of TTP. J. Chem. Theory Comput. 12: 4717-4725.
- Deveau, L. M.; Massi, F. (2016 ) Three residues make an evolutionary switch for folding and RNA-destabilizing activity in the TTP family of protein. ACS Chem. Biol. 11: 435-43.
Fabio C. L. Almeida
Federal University of Rio de Janeiro, Brazil
Title: Role of conformational equilibrium in molecular recognition and capsid assembly: the case of flavivirus capsid proteins
Time :

Biography:
Fabio C. L. Almeida has his expertise in protein structure and dynamics by nuclear magnetic resonance (NMR). He solved the structure of several proteins by NMR. He has important contribution in the structure and dynamics of plant defensins. Fabio and his group showed that despite the conserved folding, defensins display a wide variation in dynamics, which enabled the mapping of binding regions and description of the mechanism of membrane recognition. The group also showed that dynamics are also the key for the understanding of the mechanism of membrane recognition of antimicrobial peptides. Pre-existent order in flexible peptides permits discrimination between the regions of specific and non-specific binding. Fabio´s group also described the structure and dynamics of the water cavity of thioredoxin, which is an essential structural element for catalysis. Fabio is the director of the National Center of NMR (CNRMN, http://cnrmn.bioqmed.ufrj.br) and president of the Brazilian NMR Association (AUREMN).
Abstract:
Proteins are dynamic entities able to move in a wide range of timescales that goes from picoseconds to seconds. Motions that occur in microseconds to seconds define biologically relevant events that are frequently involved in binding, allostery and catalysis1,2. In our laboratory, we used relaxation parameter, relaxation dispersion experiments and molecular dynamic simulation to correlate conformational equilibrium with molecular recognition and catalysis.
Dengue and Zika are major arthropod-borne human viral disease, for which no specific treatment is available. The flavivirus capsid protein is the trigger of virus assembly. Capsid proteins are located at the cytoplasm bound to lipid droplets (LD). Binding to LDs are essential for virus assembly3,4. We previously showed that the positively charged N-terminal region of Dengue virus capsid protein prompts the interaction with negatively charged LDs, after which a conformational rearrangement enables the access of the central hydrophobic patch to the LD surface5. We also showed the participation of the intrinsically disordered region in binding and possible regulation of capsid assembly6.
We probed the structure and dynamics of Dengue virus and Zika virus capsid proteins (DENVC and ZkC) by nuclear magnetic resonance. They bind lipid droplets (LD) in the cytoplasm, which mediates virus assembly in an unknown way. We showed that the dynamics of the capsid protein is intrinsically involved in the mechanism of LD and RNA binding and virus assembly. We also measured binding to nucleic acids and probed the assembly using small angle x-ray scattering and negative staining electron microscopy. The understanding of the participation of the intrinsically disordered N-terminal region and its dynamics helped us propose a mechanism for Dengue and Zika virus assembly and to develop a peptide with the potential to block virus assembly.
ACKNOLEDGEMENTS: FAPERJ, CAPES, CNPq, INBEB-CNPq.
References:
- Iqbal, A., Moraes, A. H., Valente, A. P. & Almeida, F. C. L. Structures of the reduced and oxidized state of the mutant D24A of yeast thioredoxin 1: insights into the mechanism for the closing of the water cavity. J. Biomol. NMR 63, 417–423 (2015).
- de Paula, V. S., Razzera, G., Barreto-Bergter, E., Almeida, F. C. L. & Valente, A. P. Portrayal of complex dynamic properties of sugarcane defensin 5 by NMR: multiple motions associated with membrane interaction. Structure 19, 26–36 (2011).
- Samsa, M. M. et al. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 5, e1000632 (2009).
- Faustino, A. F. et al. Dengue virus capsid protein interacts specifically with very low-density lipoproteins. Nanomedicine 10, 247–55 (2014).
- Martins, I. C. et al. The disordered N-terminal region of dengue virus capsid protein contains a lipid droplet-binding motif. Biochem. J. 444, 405–415 (2012).
Sushant Kumar
Yale University, USA
Title: Integrative Approaches for Variant Interpretation in Coding Regions
Biography:
Sushant Kumar is a post-doctoral associate in the molecular biophysics and biochemistry department at the Yale university. He has extensive experience in biological data mining, proteins simulations and cancer genomics. He is particularly interested in integrating genomic variation data and protein structural data to develop novel methods assessing disease variant impact. In past, he has applied coarse-grained models to decipher the role of various physical factors influencing the coupled folding and binding mechanism observed among disordered proteins.
Abstract:
Statement of the Problem: The exponential rise in next-generation sequencing data is presenting considerable challenges in terms of variant interpretation. Though deep sequencing is unearthing large numbers of rare single nucleotide variants (SNVs), the rarity of these variants makes it difficult to evaluate their potential deleteriousness with conventional phenotype-genotype associations. Furthermore, many disease-associated SNVs act through mechanisms that remain poorly understood. 3D protein structures may provide valuable substrates for addressing these challenges. We present two general frameworks for doing so. In our first approach, we use localized frustration, which quantifies unfavorable residue interactions, as a metric to investigate the local effects of SNVs. In contrast to this metric, previous efforts have quantified the global impacts of SNVs on protein stability, despite the fact that local effects may impact functionality without disrupting global stability (e.g. in relation to catalysis or allostery). In our second approach, we employ models of conformational change to identify key allosteric residues by predicting essential surface pockets and information-flow bottlenecks (a new software tool that enables this analysis is also described). Importantly, although these two frameworks are fundamentally structural in nature, they are computationally efficient, thereby making analyses on large datasets accessible. We detail how these database-scale analyses shed light on signatures of conservation, as well as known disease-associated variants, including those involved in cancer.

Figure 2: The effect of introducing a typical deleterious SNV (∆F < 0). Each of the two vertical lines represents an energy-level diagram. Each level on this energy scale corresponds to the total energetic value of the protein if the residue position) were to be occupied by distinct amino acids. The ∆F associated with an SNV is negative if the SNV introduces a destabilizing effect.
References:
- Kumar S, Clarke D, Gerstein M(2016) Localized structural frustration for evaluating the impact of sequence variants. Nucleic Acid Research 429:435-445.
- Clarke D, Sethi A, Li Shantao, Kumar S, Chang RW, Chen J, Gerstein M (2016) Identifying Allosteric Hotspots with Dynamics: Application to Inter- and Intra-species Conservation. Structure 24:826-37.
- Sethi A, Clarke D, Chen J, Kumar S, Galeev TR, Regan L, Gerstein M (2015) Reads meet rotamers: structural biology in the age of deep sequencing. Current Opinion in Structural Biology 35:125-34.
Abdulrahman Alshehri
University of Sheffield, UK
Title: Generation of long acting therapies using glycosylated linkers
Biography:
Abdulrahman Alshehri working at security Forces Hospital, Riyadh, Saudi Arabia for 12 years. I did my Phd at sheffield University, UK. I have been working in creating long acting therapies using different stratigies such as, glycosylated linkers. Using multiple techniquies like PCR, gene cloning, cell culture, pharmacodynamic and pharmacokinetic to generate these therapies.
Abstract:
Rationale: The current therapeutic drugs such as, growth hormone (GH), granulocyte colony-stimulating factor (GCSF) and leptin require once-daily injections, which are inconvenient and expensive. Therefore, a number of approaches to reducing therapeutic regimens clearance have been tried mainly through conjugation with another moiety. One such technology already being employed is PEGylation; however this has been shown to be non-biodegradable and toxic. A previous study by Asterion has shown that the use of glycosylated-linkers between two GH ligands to create protein-tandems resulted in their glycosylation and an increased molecular weight (MW) whilst maintaining biological activity. The use of this technology using GCSF as an example will be presented, but can be easily applied to other molecules such as leptin.
Hypothesis: The incorporation of variable glycosylated linkers between two GCSF ligands will create a construct with high molecular weight and protected from proteolysis resulting in reduced clearance with out blocking bioactivity.
Methodology: GCSF tandems with linkers containing between 2-8 NAT glycosylation motifs and their respective controls (Q replaces N in the sequence motif NAT so there is no glycosylation) were cloned, and sequenced. Following expression in Chinese hamster ovary (CHO) cells, expressed protein was analysed by SDS-PAGE to confirm molecular weights. In vitro bioactivity was tested using an AML-193 proliferation assay. Immobilised Metal Affinity Chromatography (IMAC) was used to purify the protein. Pharmacokinetic and pharmacodynamics properties of the purified GCSF tandem proteins were measured in normal Sprague Dawley rats with full ethical approval.
Results: Purified glycosylated tandems show increased molecular weight above that of controls when analysed by SDS-PAGE. All GCSF tandems show increased bioactivity in comparison to native GCSF. Following intravenous administration to rats, GCSF2NAT, GCSF4NAT, GCSF8NAT containing 2, 4 & 8 glycosylation sites respectively and GCSF8QAT (nonâ€glycosylated GCSF tandem control) showed approximately 3â€fold longer circulating halfâ€life compared to that reported for the native GCSF (1.79 hours). Both GCSF2NAT and GCSF4NAT show a significant increase in the percentage of neutrophils over controls at 12 hours post injection. This effect however is short lived as the counts at 24+ hours are not significantly different to controls. GCSF8NAT shows an increase in the percentage of neutrophils that is only significant at 48 hours.
Conclusion: Results show that the use of glycosylated linkers to generate GCSF tandems results in molecules with increased molecular weight, improved in vitro bioactivity, longer circulating half-lives and enhanced neutrophilic population when compared to both native GCSF and the non-glycosylated tandem protein.

Figure 1: An example of 2NAT glycosylation motifs and its control 2QAT within a flexible linker (Gly4Ser)n between two GCSF ligands. (A) The glycosylation motif 2NAT inserted to the linker (glycosylated linker). (B) Non-glycosylation motif 2QAT control.
References:
- LI & D'ANJOU 2009. Pharmacological significance of glycosylation in therapeutic proteins. Curr Opin Biotechnol, 20, 678-84.
- SINCLAIR & ELLIOTT 2005. Glycoengineering: the effect of glycosylation on the properties of therapeutic proteins. J Pharm Sci, 94, 1626-35.
- TANAKA et al. 1991. Pharmacokinetics of recombinant human granulocyte colony-stimulating factor conjugated to polyethylene glycol in rats. Cancer Res, 51, 3710-4.
- CHUNG, H. K., KIM, S. W., BYUN, S. J., KO, E. M., CHUNG, H. J., WOO, J. S., YOO, J. G., LEE, H. C., YANG, B. C., KWON, M., PARK, S. B., PARK, J. K. & KIM, K. W. 2011. Enhanced biological effects of Phe140Asn, a novel human granulocyte colony-stimulating factor mutant, on HL60 cells. BMB Rep, 44, 686-91.
- COOPER, K. L., MADAN, J., WHYTE, S., STEVENSON, M. D. & AKEHURST, R. L. 2011. Granulocyte colony-stimulating factors for febrile neutropenia prophylaxis following chemotherapy: systematic review and meta-analysis. BMC Cancer, 11, 404.
- Young Researchers Forum
Location:
Session Introduction
Jessica L Thomaston
UCSF, USA
Title: XFEL structures of the M2 proton channel of influenza A reveal pH-dependent water networks under room temperature conditions

Biography:
Jessica Thomaston is a PhD candidate in the lab of Professor William DeGrado at the University of California, San Francisco. She studies the structure of the influenza M2 proton using lipidic cubic phase crystallization techniques and x-ray diffraction at synchrotron and XFEL sources. The M2 protein is among the smallest proton channels found in nature and is also a drug target against the flu. Her work focuses on the proton conduction mechanism of the M2 channel, particularly the involvement of water in proton transport and the structural characterization of how drugs and novel inhibitors bind to the channel and block proton conduction.
Abstract:
The M2 proton channel of influenza A is a drug target that is essential for replication of the flu virus. It is also a model system for the study of selective, unidirectional proton transport across a membrane. Ordered water molecules arranged in “wires” inside the channel pore have been proposed to play a role in the conduction of protons to the four gating His37 residues and the stabilization of multiple positive charges within the channel. Previous crystallographic structures determined using a synchrotron radiation source were biased by cryogenic data collection conditions, and room-temperature data collection was limited by radiation damage. These problems have been avoided through room temperature diffraction at an X-ray free electron laser (XFEL).
Data were collected at an XFEL source to a resolution of 1.4 Å at three different pH conditions: pH 5.5, pH 6.5, and pH 8.0. Here, we examine the ordering of water in the M2 pore within crystals containing only the Copen conformation, which is an intermediate that accumulates at high protonation of the His37 tetrad. This allows us to access multiple protonation states of His37 in the Copen conformation and probe changes in solvent ordering prior to and following the release of a proton into the viral interior.
At pH 5.5, a continuous hydrogen bonded network of water molecules spans the vertical length of the channel, consistent with a Grotthuss mechanism model for proton transport to the His37 tetrad. This ordered solvent at pH 5.5 could act to stabilize the positive charges that build up on the gating His37 tetrad during the proton conduction cycle. The number of ordered pore waters decreases at higher pH, where the Copen state is less stable. These studies provide a graphical view of the response of water to a change in charge within a restricted channel environment.
References:
- "XFEL structures of the M2 proton channel of influenza A reveal pH-dependent water networks under room temperature conditions." Thomaston JL, Woldeyes RA, Nakane T, Yamashita A, Tanaka T, Koiwai K, Brewster AS, Barad BA, Chen Y, Lemmin T, Uervirojnangkoorn M, Arima T, Kobayashi J, Masuda T, Suzuki M, Sugahara M, Sauter NK, Tanaka R, Nureki O, Tono K, Joti Y, Nango E, Iwata S, Yumoto F, Fraser JS, DeGrado WF. In press, PNAS July 2017.
- "Crystal structure of the drug-resistant S31N influenza M2 proton channel." Thomaston JL, DeGrado WF. Protein Science 25(8):1551-4. August 2016.
- "High resolution structures of the M2 proton channel from influenza A virus reveal dynamic pathways for proton stabilization and transduction." Thomaston JL, Alfonso-Prieto M, Woldeyes R, Fraser JS, Klein ML, Fiorin G, DeGrado WF. PNAS 112(46):14260-5. November 2015.
- "Detection of drug-induced conformational change of a transmembrane protein in lipid bilayers using site-directed spin labeling." Thomaston JL, Nguyen P, Brown EC, Upshur MA, Wang J, DeGrado WF, Howard KP. Protein Science 22(1): 65–73. January 2013.

Biography:
Maharani Pertiwi Koentjoro is a 3rd year PhD student and the Monbukagakusho fellow in the United Graduate School of Agricultural Science, Gifu University, Japan. She holds a BA in Institute of Technology “Sepuluh Nopember”, Indonesia, and a Master in Gadjah Mada University, Indonesia. Her research interests include a molecular biological and biochemical investigation on bacteria. Currently she is working on projects structural studies of complex molecular machines that initiates LysR-Type Transcription Regulator in bacteria.
Abstract:
Cupriavidus necator NH9, which can utilize chlorocatechol as a sole carbon and energy source, degrades chlorocatechol with enzymes of the ortho-cleavage pathway. These enzymes are coded in the cbnABCD operon, of which expression is specifically regulated by a LysR-type transcriptional regulator (LTTR) CbnR. CbnR forms a tetramer and can be regarded as a dimer of dimers. The tetrameric CbnR has four DNA- binding domains and these DNA-binding domains recognize approximately 60 bp DNA sequence. The binding sequence is composed of two binding sites, recognition binding site (RBS) and activation binding site (ABS). Each binding site seems to be recognized by two DNA-binding domains in the tetramer. While the crystal structure of the tetrameric CbnR has already been determined1, the molecular mechanism of DNA recognition by CbnR remains elusive. We therefore initiated the crystal structure analysis of DNA-binding domain of CbnR (CbnR(DBD)) in complex with RBS. The crystal structure would give an insight into the molecular mechanism of the CbnR-DNA interaction, which is the first step to understand the gene activation mechanism by LTTR.
Here we report the crystal structure of CbnR(DBD) (residues 1 - 87) in complex with RBS, a 25-bp DNA fragment. The crystal structure was determined by the MR-native SAD method at 2.55 Å resolution with Rwork/Rfree of 0.221/0.264. The crystal structure shows that dimeric CbnR(DBD) interacts with RBS. The dimeric CbnR(DBD) adopts essentially the same conformation as that in the tetramic CbnR with the root mean squares deviation of 1.1 Å (174 Cα atoms). The a3 helix and the winged region of the winged-helix turn helix (wHTH) motif in CbnR(DBD) directly interact with the major and minor grooves of promoter sequence, respectively, and the interactions seem to bend DNA by approximately 30°. To further analyse the molecular mechanism of their interaction, biochemical analysis is in progress.

References:
-
Muraoka, S., Okumura, R.., Uragami, Y., Nonaka, T., Ogawa, N., Miyashita, K. and Senda, T (2003) Purification and crystallization of a Lysr–type transcriptional regulator CbnR from Ralstonia eutropha NH9. Protein and Peptide Letters, 10 (3): 325–329.
-
Ogawa, N. McFall, SM., Klem, TJ., Miyashita, K and Chakrabarty, AM (1999) Transcriptional activation of the clorocatechol degradative genes of Ralstonia eutropha NH9. Journal of Bacteriology, 181 (21): 6697–6705.
-
Ruangprasert, A., Craven, SH., Neidle, EL., and Momany, C. 2010. Full–length structures of BenM and two variants reveal different oligomerization schemes for LysR–Type transcriptional regulators. Journal of Molecular Biology, 404:568–586.
-
Devese, L., Smirnova, I., Lonneborg, R., Kapp, U., Brzezinski, P., Leonard, GA. and Dian, C (2011) Crystal structures of DntR inducer binding domains in complex with salicylate offer insights into the activation of LysR–type transcriptional regulators. Molecular Microbiology, 81 (2): 354–367.
-
Alanazi, AM., Neidle, EL., and Momany, C (2013) The DNA-binding domain of BenM reveals the structural basis for the recognition of a T-N11-A sequence motif by LysR-type transcriptional regulators. Acta Crystallographica, D69: 1995-2007.
Nadia L. Opara
Paul Scherrer Institute, Switzerland
Title: Delivery methods for Free Electron Lasers: direct protein crystallization on solid supports economizes sample consumption in serial femtosecond crystallography

Biography:
Nadia Opara joined the CINA group at the Biozentrum University of Basel and the LMN at PSI in Switzerland in 2014 in the frame of SNI PhD school program to work on a project aiming at improving sample preparation methods for XFEL-based protein nano-crystallography. Beforehand she completed her bachelor studies in chemistry and a master program in molecular biotechnology at the Lodz University of Technology in Poland.
Abstract:
Classical crystallography methods based on synchrotrons usually require crystals of relatively large dimensions, i.e. above about 5 micrometers. The recent availability of X-ray free electron laser sources (XFELs) providing femtosecond X-ray pulses of ultrahigh brightness facilitate the investigation of nanocrystals. However, in this case data collection has to be performed in the mode of the serial crystallography in so-called diffraction-before-destruction regime because the probed area of the sample is completely destroyed after the interaction with ultraintense radiation. As thousands of crystals have to be provided sequentially to the XFEL beam, selection of an efficient sample delivery system is crucial to minimize protein consumption during data collection.
Delivery methods applied so far include steady streaming liquid jets [1,2] of the crystal suspension. The application of more viscous media like lipidic cubic phase (LCP) [3], agarose [4] or hyaluronic acid [5] matrices has also been demonstrated. However all these methods use significant amounts of the precious protein, which cannot be recovered even if not directly probed.
Recent developments of drop of demand methods [6] or fixed targets [1,7] allow overcoming this problem. But still, handling of the fragile crystals should be gentle or, at best, avoided.
Microfabricated silicon chips with ultrathin Si3N4 membranes provide the possibility to regularly position crystals on precisely defined spots by direct crystallization using classical vapor diffusion method [8]. The sample consumption is minimal since crystal growth takes place in nanoliter volume cavities. No additional sample transfer is needed, because X-rays are probing the crystals at the spot where they grew on the X-ray-transparent ultrathin amorphous silicon nitride membranes. Assembly with a second chip to form a hermetically sealed sandwich protects specimens from dehydration and facilitates in situ diffraction data collection at room temperature, as demonstrated in a synchrotron experiment providing high-resolution patterns [Fig. 1].
References:
- Muniyappan S et al. (2015) Recent advances and future prospects of serial crystallography using XFEL and synchrotron X-ray sources. Bio Design 3:2
- Steinke I et al. (2016) A liquid jet setup for x-ray scattering experiments on complex liquids at free-electron laser sources. Rev Sci Instrum. 87:063905
- Weierstall U et al. (2014) Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat Commun. 5:3309
- Conrad CE et al. (2015) A novel inert crystal delivery medium for serial femtosecond crystallography. IUCrJ 2:421-30
- Sugahara M et al. (2016) Oil-free hyaluronic acid matrix for serial femtosecond crystallography. Sci Rep. 6:24484
Ji Won Kim
Inha University, South Korea
Title: Induced Phenotype Targeted Chemotherapy with Checkpoint Kinase 1 Inhibitor

Biography:
Ji Won Kim has her passion in improving the health and wellbeing for head and neck cancer patients. Also she has an interest in salivary gland disease and aging. She is working in Inha university medical center after ENT resident training and fellowship in Asan Medical center. She worked as a member in asan institute for life science. She has a lot of papers about head and neck cancers treatment and therapeutic approach to overcome salivary dysfunction.
Abstract:
Backgound: We suggested a novel strategy termed induced phenotype targeted therapy (IPTT) using radiation. The aim of this study is to expand the the clinical implications of IPTT including metastatic tumors. We investigated the candidate drugs such as checkpoint kinase 1 (Chk 1) inhibitor to induce phenotype caspase-3 and found the mechanism of activating the prodrug.
Methodology & Theoretical Orientation: We designed a caspase-3 specific activatable prodrug, DEVD-S-DOX, containing doxorubicin linked to a peptide moiety (DEVD) cleavable by caspase-3 upon apoptosis. To induce apoptosis systemically in the metastatic tumor, we used a Chk 1 inhibitor, LY2603618. clonogenicity, cell cycle distribution and apoptosis were assessed in breast cancer cell lines with prodrug and LY2603618, seperatively, and combination.The in vivo antitumor activity of the caspase-3-specific activatable prodrug combined with LY2603618 was investigated in C3H/HeN tumor-bearing mice (n = 5 per group) and analyzed with the Student's t test or Mann-Whitney U test. All statistical tests were two-sided. We confirmed the basic principle using a caspase-sensitive nanoprobe (Apo-NP).
Findings: The in vitro studies showed that the cell cycle arrest of p53-deficient cancer cells by Chk1 inhibitor, thus leading to facilitated caspase-3 upon apoptosis. The growth of MDA-MB-231, which is p53-deficient, was effectively suppressed by daily oral administration of LY2603618 when treated with DEVD-S-DOX in vivo without evident toxicities.
Conclusion & Significance: The combination therapy using DEVD-S-DOX and Chk1 inhibitor could effectively treat p53-deficient breast cancer by selectively chemosensitizing cancer cells with low toxic adverse effects.
References:
- Lee BS, Cho YW, Kim GC, Lee DH, Kim CJ, Kil HS, Chi DY, Byun Y, Yuk SH, Kim K, Kim IS, Kwon IC, Kim SY. Induced phenotype targeted therapy: radiation-induced apoptosis-targeted chemotherapy. J Natl Cancer Inst. 2014 Dec 12;107(2).
- Chung SW, Choi JU, Lee BS, Byun J, Jeon OC, Kim SW, Kim IS, Kim SY, Byun Y. Albumin-binding caspase-cleavable prodrug that is selectively activated in radiation exposed local tumor. Biomaterials. 2016 Jul;94:1-8.
- Chung SW, Kim GC, Kweon S, Lee H, Chang HW, Kim JW, Son WC, Kim SY, Byun Y. Metronomic oral doxorubicin with combination of Chk1 inhibitor for the treatment of p53-deficient breast cancer. Mol Cancer Ther.submitted

Biography:
Stephanie Morais performs her pHD under Tatiana Souza supervision and coordinate projects involving advances in leukemia treatment advances. She is part of the team since 2013 and has expertise not only in molecular and structural biology but also in cancer biology.
Abstract:
Acute Lymphoid Leukemia (ALL) is the most common neoplasia in childhood. The multi-therapeutic treatment resulted in remarkable advances in treatment of children, with 90.4% survival rate. L-asparaginase has been a central component of ALL therapy for over 40 years and acts by depleting plasma asparagine. In contrast to the normal cells, tumor cells lack the ability to synthesize asparagine and thus depend on external uptake of this amino acid for growth. Nowadays, three asparaginases are used in therapy: native L-asparaginase II from Escherichia coli, a pegylated form of this enzyme and L-asparaginase isolated from Erwinia chrysanthemi. Among the commercially available L-asparaginases, the E. coli enzyme presents the highest catalytic activity but also the highest toxicity, due to its further ability to hydrolyze glutamine, generating glutamate. Moreover, the immune response in patients under therapy with bacterial asparaginases can result in enzyme neutralization and the need to proceed the treatment with one of the alternative L-asparaginases. Based on the analysis of the available crystal structures we have designed, produced and crystallized E. coli asparaginase with modifications. Crystals diffracted up to 1.65 Å resolution at the Soleil Synchrotron. We combine structural analysis with kinetic and cellular approaches in order to identify the determinants of E. coli asparaginase toxicity. In addition, we have been working on the production of modified human asparaginases for structural characterization, kinetic and anti-leukemic activity assays. The introduction of human asparaginase in ALL treatment would avoid the problems caused by the bacterial enzymes, however a major difficulty in the therapeutic use of the human enzyme comes from the fact that human asparaginases need to undergo activation through an auto-cleavage step, which was shown to be a low efficiency process in vitro, reducing the enzyme activity. These structural analysis gather insights about how engineering asparaginases can improve ALL treatment.
References:
- MORAIS, S. B. ; WEILER, A. V. P. ; MURAKAMI, M. T. ; Souza, T. A. C. B. Characterization of Alanine Aminotransferase from Trypanosoma cruzi. Protein & Peptide Letters, 2016
- SANTOS, FRED LUCIANO NEVES ; CELEDON, PAOLA ALEJANDRA FIORANI ; ZANCHIN, NILSON IVO TONIN ; Brasil, tatiana de arruda campos ; FOTI, LEONARDO ; SOUZA, WAYNER VIEIRA DE ; SILVA, EDMILSON DOMINGOS ; GOMES, YARA DE MIRANDA ; KRIEGER, Marco Aurélio . Performance Assessment of Four Chimeric Trypanosoma cruzi Antigens Based on Antigen-Antibody Detection for Diagnosis of Chronic Chagas Disease. Plos One, v. 11, p. e0161100, 2016.
- SANTANA, ALINE GUIMARÃES ; GRACHER, ANA HELENA PEREIRA ; RÜDIGER, ANDRÉ LUIS ; ZANCHIN, NILSON IVO TONIN ; CARVALHO, PAULO COSTA ; CIPRIANI, THALES RICARDO ; DE ARRUDA CAMPOS BRASIL DE SOUZA, TATIANA . Identification of potential targets for an anticoagulant pectin. Journal of Proteomics (Print), v. 1, p. 1-4, 2016.
- MORAES, EDUARDO ; MEIRELLES, GABRIELA ; HONORATO, RODRIGO ; DE SOUZA, Tatiana ; DE SOUZA, EDMARCIA ; MURAKAMI, MARIO ; DE OLIVEIRA, PAULO ; Kobarg, Jörg . Kinase Inhibitor Profile for Human Nek1, Nek6, and Nek7 and Analysis of the Structural Basis for Inhibitor Specificity. Molecules (Basel. Online), v. 20, p. 1176-1191, 2015.
- DOMINGUES, MARIANE NORONHA ; SFORÇA, MAURICIO LUIS ; SOPRANO, ADRIANA SANTOS ; LEE, JACK ; DE SOUZA, TATIANA DE ARRUDA CAMPOS BRASIL ; CASSAGO, ALEXANDRE ; PORTUGAL, RODRIGO VILLARES ; DE MATTOS ZERI, ANA CAROLINA ; MURAKAMI, MARIO TYAGO ; SADANANDOM, ARI ; DE OLIVEIRA, PAULO SERGIO LOPES ; BENEDETTI, CELSO EDUARDO . Structure and Mechanism of Dimer-Monomer Transition of a Plant Poly(A)-Binding Protein upon RNA Interaction: Insights into Its Poly(A) Tail Assembly. Journal of Molecular Biology, v. 427, p. 2491-2506, 2015.
- Special Session: Structural Biology & Single Molecules
Chair
Yuri L Lyubchenko
University of Nebraska Medical Center, USA
Session Introduction
Yuri L. Lyubchenko
University of Nebraska Medical Center, USA
Title: Nanoscale structure and dynamics of centromere nucleosomes
Time : 10:50-11:10

Biography:
Yuri L. Lyubchenko is Professor of Pharmaceutical Sciences at the University of Nebraska Medical Center, Omaha, NE, USA. His research focuses on understanding fundamental mechanisms underlying health and disease, which are key to developing new and more effective diagnostics and medications. This primarily basic research allows him not only identify new drug targets for small molecule drugs, it also leads to development of the nanotools and methods to discover novel approaches for diagnostic, treatment and disease prevention and to more rapidly determine their efficacy at the molecular level.
Abstract:
Statement of the Problem: Chromatin integrity is crucial for normal cell development. The cell division process is accompanied by the segregation of replicated chromosome, and chromatin centromeres, specialized segments of chromosomes provide the accuracy of the chromosomal segregation. If the centromere becomes damaged or removed, chromosomes segregate randomly disrupting the cell division process. The centromeres are specifically recognized by kinetochores suggesting that centromeres contain specific structural characteristics. However, these structural details and the mechanism underlying their highly specific recognition remain uncertain. Methodology & Theoretical Orientation: In this study, we performed direct imaging of CENP-A nucleosome core particles by time-lapse high-speed atomic force microscopy (AFM), enabling us to directly visualize the dynamics of CENP-A nucleosomes. Nucleosomes used for evaluation of DNA wrapping around the histone core were assembled on a DNA substrate containing a centrally positioned 601 motif. Findings: A broadly dynamic behavior of the DNA flanks was first revealed by analysis of AFM images acquired in ambient conditions. Time-lapse imaging further identified the distinctive pathways unique to CENP-A-nucleosome dynamics that are not shared by H3. The spontaneous unwrapping of DNA flanks can be accompanied by the reversible and dynamic formation of loops with sizes equivalent to a single wrap of DNA. Translocation of CENP-A nucleosomes was observed, with the formation of internal DNA loops along the nucleosome. This process was reversible, settling the core back to its starting position. Additionally, the transfer of the histone core from one DNA substrate to another was visualized, as well as distinctive splitting into sub-nucleosomal particles that was also reversible. Conclusion & Significance: Altogether, our data suggest that unlike H3, CENP-A is very dynamic, permitting its nucleosome to distort freely and reversibly, which in turn allows a longer term stability, which may play a critical role in centromere integrity during mitosis and replication.
References:
[1] Lyubchenko, Y. L. (2014) Centromere chromatin: a loose grip on the nucleosome?, Nat Struct Mol Biol 21, 8.
[2] Lyubchenko, Y. L., and Shlyakhtenko, L. S. (2015) Chromatin imaging with time-lapse atomic force microscopy, Methods Mol Biol 1288, 27-42.
[3] Lyubchenko, Y. L., and Shlyakhtenko, L. S. (2015) Atomic Force Microscopy Imaging and Probing of Amyloid Nanoaggregates, In Handbook of Clinical Nanomedicine: From Bench to Bedside (Bawa, R., Audette, G. & Rubinstein, I., Ed.), p 1500. , Pan Stanford Publishing, Singapore. .
[4] Sun, Z., Tan, H. Y., Bianco, P. R., and Lyubchenko, Y. L. (2015) Remodeling of RecG Helicase at the DNA Replication Fork by SSB Protein, Sci Rep 5, 9625.
[5] Proctor, E. A., Fee, L., Tao, Y., Redler, R. L., Fay, J. M., Zhang, Y., Lv, Z., Mercer, I. P., Deshmukh, M., Lyubchenko, Y. L., and Dokholyan, N. V. (2016) Nonnative SOD1 trimer is toxic to motor neurons in a model of amyotrophic lateral sclerosis, Proc Natl Acad Sci U S A 113, 614-619.
John van Noort
Leiden University, The Netherlands
Title: The structure of chromatin; single-molecule experiments on model fibers and real genes

Biography:
Chromatin is the ubiquitous protein-DNA complex that forms the structural basis of DNA condensation in all eukaryotic organisms. Packaging and depackaging of chromatin, called chromatin remodeling, plays a central role in all cellular processes that involve chromosomes such as transcription, replication, recombination and repair. Detailed knowledge of the principles and mechanisms underlying this control of DNA condensation is thus vital for understanding many diseases, including neurological disorders and cancer. The physical mechanisms governing these processes however, are still largely unknown. I am interested in developing and using modern biophysical techniques to unravel the physics behind DNA condensation and its role in transcription regulation.
Abstract:
The folding of chromatin defines access to our genes and therefore plays a pivotal role in transcription regulation. However, the structure of chromatin fibers is poorly defined and heavily debated. We used single-molecule techniques to probe and manipulate the dynamics of nucleosomes in individual chromatin fibers. These novel methods were initially applied to synthetic, highly homogeneous nucleosomal arrays and yielded unprecedented insight in the structure and dynamics of chromatin.
With single pair Forster Resonance Energy Transfer we showed that the nucleosome is very dynamic, unwrapping half of its DNA four times per second. Using single molecule force spectroscopy, it was possible to measure the kinetics of this unfolding, both in single nucleosomes and in well-defined arrays of nucleosomes that fold into a 30 nm fiber. Analysis of the unfolding pattern reveals a linker length dependence of the higher order folding.
The linker length in vivo however varies, and to obtain insight the positioning of nucleosomes we developed a simple statistical physics model that captures sequence dependent positioning effects for both reconstitutions on synthetic DNA and chromatin in vivo.
We recently developed a method to purify specific chromatin fragments from yeast without crosslinking the fiber while maintaining the complexity that provides functionality to our epi-genome. I will show the first single-molecule force spectroscopy results on intact, native fibers which uniquely probe chromatin structure, composition and variations in it at the single-molecule level.
Refernces:
-
Multiplexing genetic and nucleosome positioning codes: a computational approach (2016) B Eslami-Mossallam, RD Schram, M Tompitak, J van Noort, H Schiessel PloS one 11 (6), e0156905
-
Quantitative analysis of single-molecule force spectroscopy on folded chromatin fibers (2016) H Meng, K Andresen, J Van Noort Nucleic acids research 43 (7), 3578-3590
-
spFRET reveals changes in nucleosome breathing by neighboring nucleosomes (2015) R Buning, W Kropff, K Martens, J van Noort Journal of Physics: Condensed Matter 27 (6), 064103
-
Histone H3 phosphorylation near the nucleosome dyad alters chromatin structure (2014) Justin A North, Marek Šimon, Michelle B Ferdinand, Matthew A Shoffner, Jonathan W Picking, Cecil J Howard, Alex M Mooney, John van Noort, Michael G Poirier, Jennifer J Ottesen Nucleic acids research 42 (8), 4922-4933
-
Sequence-based prediction of single nucleosome positioning and genome-wide nucleosome occupancy (2012) T van der Heijden, JJFA van Vugt, C Logie, J van Noort Proceedings of the National Academy of Sciences 109 (38), E2514-E25225.
Peter Hinterdorfer
Johannes Kepler University Linz , Austria
Title: Deciphering and filming molecular recognition at the nano-scale with AFM
Time :

Biography:
Peter Hinterdorfer performs advanced nanoscopic techniques in nano-bio technology, life science, and medical diagnostics, and has been working on antibody/antigen interactions, transmembrane transporters, virus/membrane interactions, cells of the immune system, nuclear envelope membranes, and bacterial surface layers. He has done pioneering work in single molecule force spectroscopy and has invented a combined topography and recognition imaging technique. Recently he did research with high-speed bio-AFM.
Abstract:
In molecular recognition force microscopy (MRFM), ligands are covalently attached to atomic force microscopy tips for the molecular recognition of their cognitive receptors on probe surfaces1. Interaction forces between single receptor-ligand pairs are measured in force-distance cycles. The dynamics of the experiment is varied2, which gives insight into the molecular dynamics of the receptor-ligand recognition process and yields information about the binding pocket, binding energy barriers, and kinetic reaction rates3. Combination of high-resolution atomic force microscope topography imaging with single molecule force spectroscopy provides a unique possibility for the localization of specific molecular recognition events4. The identification and visualization of receptor binding sites on complex heterogeneous bio-surfaces such as cells and membranes are of particular interest in this context4. Considered as the paradigm for molecular recognition are antibodies. They are key molecules for the immune system of vertebrates. The Y-shaped antibody type IgG exhibits C2-symmetry; its Fc stem is connected to two identical Fab arms, binding antigens by acting as molecular callipers. Bivalent binding of the two Fab arms to adjacent antigens can only occur within a distance of roughly 6 to 12 nm. AFM cantilevers adorned with an antibody can measure the distances between 5-methylcytidine bases in individual DNA strands with a resolution of 4Å, thereby revealing the DNA methylation pattern6, which has an important role in the epigenetic control of gene expression. Moreover, due to their nano-mechanical properties antibodies exhibit “bipedal” walking on antigenic surfaces7. The walking speed depends on the lateral spacing and symmetry of the antigens. Importantly, the collision between randomly walking antibodies was seen to reduce their motional freedom. It leads to formation of transient assemblies, which are known to be nucleation sites for docking of the complement system and/or phagocytes as an important initial step in the immune cascade.
References:
- Hinterdorfer, P. et al. Proc. Natl. Acad. Sci. USA 93, 3477 (1996)
- Hinterdorfer, P. et al. Nature Methods 5, 347 (2006)
- Kienberger, F. et al. Acc. Chem. Res. 39, 29 (2006)
- Preiner, J. et al. Nanotechnology 20, 215103 (2009)
- Chtcheglova, L.A. et al. Biophys J. 93, L11 (2007)
Marco Capitanio
University of Florence, Italy
Title: High-speed optical tweezers for the study of protein-DNA interaction

Biography:
Marco Capitanio is Senior Researcher at the Department of Physics of the University of Florence, Italy, and Group Leader at the European Laboratory for Non-linear Spectroscopy (LENS). After finishing his master’s degree in physics, he gave in to his long- standing interest in biology and obtained his PhD in physiology.
He then joined LENS, a research institute which is part of a European network of laser and spectroscopy facilities.
His research interests lie across physics and biology. On one hand, his research is focused on the physics of biological systems and on the development of techniques for the study of biology at the molecular scale, with a particular interest on optical methods. On the other hand, he is particularly interested in the molecular mechanisms underlying mechanical regulation of biological systems and the conversion of mechanical signals into changes in gene expression and cell fate.
Abstract:
We developed a constant-force laser trap that allows us to investigate molecular interactions and sub-nanometer conformational changes occurring on a time scale of few tens of microseconds. [Capitanio et al., Nature Methods 9, 1013-1019 (2012)]. The method is effective in studying the sequence-dependent affinity of DNA-binding proteins along a single DNA molecule. The improvement in time resolution provides important means of investigation on the long-puzzled mechanism of target search on DNA. In fact, one poorly understood issue in the field of protein-DNA interaction is how proteins weakly interact with non-cognate DNA sequences and how they efficiently find the sequence of interest among an extremely large amount of non-specific sequences. Using our technique, we could discriminate sequence and conformational -dependent interactions of a single Lac repressor protein (LacI) on DNA at physiological salt concentrations. The lac operon is a well-known example of gene expression regulation, based on the specific interaction of LacI with its cognate DNA sequence (operator). We observed LacI switching between different interaction modalities on DNA (weak, strong, sliding), depending on the molecule conformation and DNA sequence. We provide a method for measuring 1D-diffusion constants of DNA-binding proteins along DNA with a spatial resolution of about 30 base pairs, observing a broad distribution of 1D-diffusion constants of LacI and sequence-dependent diffusion constants. Our measurements provide a model of target-search and molecular switching mechanism of Lac repressor.
References:
- Gardini, L., Capitanio, M., Pavone F.S., “3D tracking of single nanoparticles and quantum dots in living cells by out-of-focus imaging with diffraction pattern recognition”, Sci. Rep. 5, 16088; doi: 10.1038/srep16088 (2015).
- Monico, C., Belcastro, G., Vanzi, F., Pavone, F.S. and M. Capitanio, “Combining single-molecule manipulation and imaging for the study of protein-DNA interactions”, J. Vis. Exp. 90, e51446, doi:10.3791/51446 (2014).
- Capitanio, M and Pavone, F.S. “Interrogating biology with force: single-molecule high-resolution measurements with optical tweezers”, Biophys. J. 105, 1293-1303 (2013).
- Monico, C., Capitanio, M., Belcastro, G., Vanzi, F., Pavone, F.S., “Optical Methods to Study Protein-DNA Interactions in Vitro and in Living Cells at the Single-Molecule Level”, Int. J. Mol. Sci. 14, 3961-3992 (2013).
- Capitanio, M., Canepari, M., Maffei, M., Beneventi, D., Monico, C., Vanzi, F., Bottinelli, R. and Pavone F.S. “Ultrafast force-clamp spectroscopy of single molecules reveals load dependence of myosin working stroke”, Nature Methods, 9, 1013–1019 (2012).
Gijs Wuite
Vrije Universiteit Amsterdam, The Netherlands
Title: Sliding sleeves of XRCC4–XLF bridge DNA and connect fragments of broken DNA

Biography:
Gijs Wuite obtained his PhD in biophysics in 2000. Since 2001 he leads his own group at the VU University Amsterdam and in 2009 was appointed to full professor. In his research he has successfully applied quantitative physical tools to investigate fundamental problems in biology, and to search for the unification of apparently unrelated biological phenomena. Moreover, he has been at the front of recent new and fast developments of biophysical techniques that have enabled visualization, manipulation and control of complex biological reactions. Based on this research work he founded in 2014 a company (LUMICKS) that sell the technology he and his group has developed. His work has appeared in journal such as Nature, Science, PNAS and Physical Review Letters. His research has been awarded with the prestigious personal VIDI, VICI and ERC grants. In 2009 Wuite was appointed member of the Young Academy, an independent platform of young top scientists within the Royal Netherlands Academy of Arts and Sciences.
Abstract:
Non-homologous end joining (NHEJ) is the primary pathway for repairing DNA double-strand breaks (DSBs) in mammalian cells1 . Such breaks are formed, for example, during gene-segment rearrangements in the adaptive immune system or by cancer therapeutic agents. Although the core components of the NHEJ machinery are known, it has remained difficult to assess the specific roles of these components and the dynamics of bringing and holding the fragments of broken DNA together. The structurally similar XRCC4 and XLF proteins are proposed to assemble as highly dynamic filaments at (or near) DSBs2 . Here we show, using dualand quadruple-trap optical tweezers combined with fluorescence microscopy, how human XRCC4, XLF and XRCC4–XLF complexes interact with DNA in real time. We find that XLF stimulates the binding of XRCC4 to DNA, forming heteromeric complexes that diffuse swiftly along the DNA. Moreover, we find that XRCC4–XLF complexes robustly bridge two independent DNA molecules and that these bridges are able to slide along the DNA. These observations suggest that XRCC4–XLF complexes form mobile sleeve-like structures around DNA that can reconnect the broken ends very rapidly and hold them together. Understanding the dynamics and regulation of this mechanism will lead to clarification of how NHEJ proteins are involved in generating chromosomal translocations (Brouwer et al., Nature, doi:10.1038/nature18643 , 2016)
References:
- Ineke Brouwer, Gerrit Sitters, Andrea Candelli, … , Mauro Modesti, Erwin J.G. Peterman, Gijs J.L. Wuite (2016) Sliding sleeves of XRCC4-XLF bridge DNA and connect fragments of broken DNA NATURE Volume:535 Issue: 7613
- Onno D. Broekmans, Graeme A. King, Greg J. Stephens, and Gijs J.L. Wuite (2016) DNA twist stability changes with magnesium(2+) concentration PHYS. REV. LETT. Volume: 116 Issue: 25
- King, Graeme A; Peterman, Erwin J G; Wuite, Gijs J L (2016) Unravelling the structural plasticity of stretched DNA under torsional constraint NATURE COMMUNICATIONS Volume: 7 Pages: 11810
- Biebricher, Andreas S.; Heller, Iddo; … Peterman, Erwin J. G.; Wuite, Gijs J. L. (2015) The impact of DNA intercalators on DNA and DNA-processing enzymes elucidated through force-dependent binding kinetics NATURE COMMUNICATIONS Volume: 6 Article: 7304
- I Heller, G Sitters, OD Broekmans, G Farge, C Menges, W Wende, SW Hell, EJG Peterman & GJL Wuite. (2013) STED nanoscopy combined with optical tweezers reveals protein dynamics on densely covered DNA Nature Methods, 10.1038/nmeth.2599

Biography:
Dmitrii V Shalashilin is a Professor of Computational Chemistry at the University of Leeds. His research is focused on the development of efficient computational techniques for quantum and classical simulations in chemistry and their applications. The recently developed Boxed Molecular Dynamics method has a wealth of applications to structural biology and dynamics of biological molecules.
Abstract:
New applications of Boxed Dynamics (BXD) [1.2], an efficient technique to extend the time scale of molecular dynamics and simulate rare events, will be presented. BXD allows analysis of thermodynamics and kinetics in complicated molecular systems. It is a fully atomistic multiscale technique, in which thermodynamics and long-time dynamics are recovered from a set of short-time molecular dynamics simulations. BXD is many orders of magnitude faster than standard MD and can produce well converged results. Previously BXD has been applied to peptide cyclization, solution-phase organic reaction dynamics, and desorption of ions from self-assembled monolayers (SAMs) [3]. Here two new applications of BXD will be reported. First atomistic simulations of protein pulling with Atomic Force Microscope AFM) will be presented, where BXD is able to reproduce correctly the Potential of Mean Force (PMF) of a protein pulled in AFM experiments, the experimentally observed force profile and its relationship with the protein structure [4] (see Fig.1). Second, an application of BXD to enzymatic peptide cyclization will also be presented, where BXD predicts correctly the cyclizable peptide sequences [5]. All such sequences have a conformation with their C and N termini close to each other as shown at the Fig.2. In both applications calculations were done with standard force field without any adjustment of the force field parameters. Thus, BXD proves to be a good predictive tool. It is implemented in CHARMM molecular dynamics code and can be used for many other applications

Fig.1 Potential of mean force as a function of end-to-end distance calculated with BXD correlates with the structures of the unfolding protein.

Fig.2 PMF as a function of end-to-end distance for two peptides P18 and P17. Only P18, which has a stable conformation with C and N termini close to each other, is cyclizable.
References:
- Glowacki, DR; Paci, E; Shalashilin, DV (2009) Boxed Molecular Dynamics: A Simple and General Technique for Accelerating Rare Event Kinetics and Mapping Free Energy in Large Molecular Systems. Journal of Physical Chemistry 113: 16603-16611.
- Shalashilin, DV; Beddard, GS; Paci, E; Glowacki, DR (2012) Peptide kinetics from picoseconds to microseconds using boxed molecular dynamics: Power law rate coefficients in cyclisation reactions. Journal of Chemical Physics 137: 165102.
- Booth, JJ; Vazquez, S; Martinez-Nunez, E; Marks, A; Rodgers, J; Glowacki, DR; Shalashilin DV (2014) Recent applications of boxed molecular dynamics: a simple multiscale technique for atomistic simulations. Philosophical Transactions of the Royal Society A - Mathematical Physical and Engineering, 372: 20130384.
- Booth, JJ; Shalashilin, DV, (2016) Fully Atomistic Simulations of Protein Unfolding in Low Speed Atomic Force Microscope and Force Clamp Experiments with the Help of Boxed Molecular Dynamics. Journal of Physical Chemistry 120: 700-708.
- Booth, JJ; Alexandra-Crivac, CN; Rickaby, KA; Nneoyiegbe, AF; Umeobika, U; McEwan, AR;
- Track 1: Recent Advances in Structural Biology
Session Introduction
Nebojsa Janjic
SomaLogic, Inc., U.S.A.
Title: Structural insights from aptamers with base modifications
Time :

Biography:
Nebojsa Janjic has been Chief Science Officer at SomaLogic, Inc. since January 2009. Prior to joining SomaLogic, Dr. Janjic was a founder and CSO at Replidyne, Inc., a biotechnology company focusing on the development of new small-molecule antibacterial agents. Prior to Replidyne, Dr. Janjic was senior director of drug discovery at NeXstar Pharmaceuticals, where his contributions include the discovery and early development of Macugen, the first aptamer to receive FDA approval and the first VEGF inhibitor developed for the treatment for macular degeneration. As CSO at SomaLogic, Dr. Janjic is involved in developing a new generation of modified aptamers and identifying opportunities for their use in science and medicine. Dr. Janjic received his bachelor's degree in molecular biology and Ph.D. in physical organic chemistry from the University of Washington in Seattle and completed his postdoctoral training at the Scripps Research Institute in La Jolla as a Cancer Research Institute Fellow.
Abstract:
Statement of the Problem: The ability to fold into distinct three-dimensional structures is the basis of high affinity and specificity characteristic of aptamer binding to their targets. We have recently introduced base modifications that increase chemical diversity of functional groups front-loaded in randomized nucleic acid libraries from which aptamers are selected. Such modifications have allowed us to identify high-affinity aptamers to a large number of protein targets previously considered “difficult” with conventional nucleic acid libraries. At the same time, our ability to predict the structures of modified aptamers with conventional nucleic acid folding rules was severely compromised, suggesting new rules for folding. Methodology & Theoretical Orientation: We examined published structures of sixteen aptamers co-crystallized with their protein targets, including three aptamers with base modifications we reported recently. Findings: In contrast to small molecules, which are entirely encaged by aptamers, proteins present large surfaces with distinct features that are recognized by complementary surfaces on aptamers. The size of these interaction surfaces is comparable to those observed with antibodies, although for aptamers, the size range is wider on both small and large extremes. The highly flexible phosphodiester backbone allows assembly of known as well as novel nucleic acid motifs into precise three-dimensional structures that orient often discontiguous aptamer regions toward their protein targets in a manner that creates surfaces with exquisite shape complementarity. Base modifications with hydrophobic side chains allow occupancy of distinctly hydrophobic pockets on proteins and create novel structural elements that illustrate the profound role modified nucleotides play in both folding and binding. Conclusion & Significance: These observations provide compelling structural rationale for the observed high affinity and specificity with which aptamers recognize their protein targets, and show us that the lexicon of structural features accessible to nucleic acid ligands can be vastly expanded with chemical modifications of nucleic acid libraries.
References:
- Davies DR, Gelinas AD, Zhang C, Rohloff JC et al. (2012) Unique motifs and hydrophobic interactions shape the binding of modified DNA ligands to protein targets. Proc Natl Acad Sci U S A 109:19971-19976.
- Gelinas AD, Davies DR, Edwards TE, Rohloff JC et al. (2014) Crystal structure of interleukin-6 in complex with a modified nucleic acid ligand. J Biol Chem 289:8720-8734.
- Jarvis TC, Davies DR, Hisaminato A, Resnicow DI et al. (2015) Non-helical DNA Triplex Forms a Unique Aptamer Scaffold for High Affinity Recognition of Nerve Growth Factor. Structure 23:1293-1304.
- Rohloff JC, Gelinas AD, Jarvis TC, Ochsner UA et al. (2014) Nucleic Acid Ligands With Protein-like Side Chains: Modified Aptamers and Their Use as Diagnostic and Therapeutic Agents. Mol Ther Nucleic Acids 3:e201.
- Gelinas, AD, Davies, DR, Janjic, N (2016) Embracing Proteins: Structural Themes In Aptamer:Protein Complexes. Curr. Rev. Struct. Biol. 36:122-132.

Biography:
Robert Craigie is a Senior Investigator in the National Institute of Diabetes and Digestive and Kidney Diseases at the National Institutes of Health, Bethesda, MD, USA. His research has focused on the mechanism of retroviral DNA and the structure and function of proteins and nucleoprotein complexes that mediate it.
Abstract:
Statement of the problem. Integration of retroviral DNA into host DNA is an essential step in the replication of HIV-1 and other retroviruses. Integration is mediated by a nucleoprotein complex (intasome) comprising the virally encoded integrase enzyme and a pair of viral DNA ends. The first intasome on the integration pathway is the stable synaptic complex (SSC) in which a pair of viral DNA ends is bridged by integrase. Within the SSC, integrase then cleaves two nucleotides from the 3’ ends of the viral DNA to form the cleaved stable synaptic complex (cSSC). The cSSC captures a target DNA and a pair of transesterification reactions covalently joins viral to target DNA. Currently approved inhibitors of HIV-1 DNA integration target intasomes (specifically the cSSC) rather than free integrase protein, High-resolution structures of intasomes are required to understand their detailed mechanism of action and how HIV-1 can escape by acquiring resistance. Methodology and strategy: Although the structures of the individual domains of HIV-1 integrase were determined more than two decades ago, attempts to obtain high-resolution structures of HIV-1 intasomes were unsuccessful. The main obstacles were the the propensity of both integrase and intasomes to aggregate and the low efficiency of assembly in vitro. We have overcome these problems by developing a hyperactive integrase mutant that assembles intasomes that are amenable to biophysical and structural studies. CryoEM studies of STCs reveal both tetrameric and higher order species that both share a common core architecture with intasomes of related retroviruses. SSCs also assemble both tetrameric and higher order intasomes and both are active for concerted DNA integration in vitro. Conclusions and significance: The results highlight how a common core intasome architecture can be assembled in different ways. Structures of cSSC intasomes in complex with inhibitors will elucidate their detailed mechanism of action and mechanisms by which HIV-1 can evolve drug resistance.
References:
- Passos D., Li M., Yang R., Rebensburg S., Ghirlando R., Jeon Y., Shkriabai N., Kvaratskhelia M., Craigie R., Lyumkis D. (2017) CryoEM Structures and Atomic Models of the HIV-1 Strand Transfer Complex Intasome. Science, 355(6320), 89-92.
- Li, M., Jurado, K.A., Lin, S., Engelman, A., and Craigie R. (2014) Engineered hyperactive integrase for concerted HIV-1 DNA Integration. PLoS ONE Volume: 9 Issue: 8 Article Number: e105078.LiX,
- Yin, Z., Lapkouski, M., Yang, W., and Craigie, R. (2012) Assembly of prototype foamy virus strand transfer complexes on product DNA bypassing catalysis of integration. Protein Science 12, 1849-1857.
- Kotova, S., Li, M., Dimitriadis, E.K., and Craigie, R. (2010) Nucleoprotein intermediates in HIV-1 DNA integration visualized by atomic force microscopy. J. Mol. Biol. 399, 491-500.
- Li, M., Mizuuchi, M., Burke, T.R., and Craigie. (2006) Retroviral DNA integration: reaction pathway and critical intermediates. EMBO. J. 25, 1295-1304.
Dino Moras
IGBMC, Strasbourg, France
Title: Allosteric control of transcription regulation by nuclear receptors: an integrative structural biology approach

Biography:
Dino Moras, PI, graduated in chemistry at the University of Strasbourg. While post-doc with M.G. Rossmann he contributed to the concept of nucleotide binding domain known as the 'Rossmann fold'. His main scientific contributions are in structural biology, related to the expression of the genetic information: (i) translation of the genetic code by aminoacyl-tRNA synthetases: discovery of the partition of aaRS in two classes and first crystal structure of a class II tRNA-aaRS complex (ii) transcription regulation by Nuclear Receptors: the first crystal structures of the ligand binding domains of two NRs (RXR and RAR) in their apo and liganded form respectively. Presently his main focus is on the molecular mechanisms of regulation using integrative structural biology approaches.
Abstract:
Nuclear Hormone Receptors interact with corepressors, coactivators and other protein cofactors to regulate signal transduction of the basal transcriptional machinery. Most NRs are known to function as dimers and with the exception of the group of oxosteroid receptors (AR, GR, MR, PR) all structural data point to a conserved interface for the ligand binding domains (LBDs) dimers.
Allosteric mechanisms control the sequential and ordered binding of nuclear receptors to the various protein effectors and target DNA. The binding of ligands induce structural transitions in the LBDs leading to the release of the corepressors and their replacement by cofactors. The LBD swallows the ligand and shields it from the solvent by closing the pocket with the C-terminal peptide. The agonist/antagonist character of the ligand is then essentially controlled by the position of helix H12 and the stability of the complex. Ligands are modulators of the activation process, their potency being defined by the fraction of time spent in the active conformation. Crystal structures of DNA binding domains (DBDs) bound to different response elements also support the proposal of DNA being an allosteric effector. The architectures of full length receptors bound to DNA fragments and cofactors have been determined by crystallography and in solution using integrative approaches. The later combine structural small angle diffraction methods by X-Rays (SAXS) and neutrons (SANS), optical techniques like FRET with labelled molecules and single particle electron microscopy (cryo-EM). Some common features emerge that rationalize the key role of DNA. The recent advances in cryo-EM allow solution structures determination at near atomic resolution. Conformational equilibrium of NRs in complex with various cofactors are also accessible.

Biography:
John (Ioannis) Vakonakis did a PhD in Biochemistry at Texas A&M University, where he pioneered the structural analysis of bacterial circadian clock proteins. His postdoctoral work at the University of Oxford focused on the structural mechanisms underpinning cell adhesion and assembly of the extracellular matrix in animals. John did breakthrough work on the molecular architecture of the centriole organelle during a second postdoc at the Swiss Light Source, prior to starting his own lab in Oxford Biochemistry. He has been a Marie Currie Fellow, Junior Research Fellow at Trinity College, Oxford, and a Wellcome Trust Research Fellow. John is now Associate Professor in Structural Biology and Biophysics at the University of Oxford, and Fellow in Biochemistry at Lincoln College. Over the last six years his research aims to understand how large molecular machines form in cells, such as the cytoadherence assemblies created upon P. falciparum-infection of human erythrocytes.
Abstract:
Human red blood cells infected by the malaria parasite Plasmodium falciparum (iRBC) form dome-shaped ~120 nm-diameter protrusions on their surface, known as ‘knobs’. Knobs provide essential presentation platforms for the parasite cytoadherence receptor family PfEMP1, which binds ligands on endothelial cells of the blood vessel wall thereby immobilizing iRBC in the microvasculature. The resulting obstruction of blood vessels and disruption of normal circulation causes inflammation and tissue damage that can lead to coma and death. iRBC cytoadherence constitutes the primary mechanism driving morbidity and mortality in P. falciparum infections, which account for over 90% of all malaria-related deaths.
Despite their importance in malaria pathology the molecular mechanisms underpinning knob formation remain poorly understood. Here, I review recent progress in characterizing knob complexes formed between parasite and parasite – host proteins. Extensive flexibility is common among parasite knob components, which necessitated an integrative approach to resolve these complexes. In particular, I will focus on the development of novel in silico docking tools suitable for evaluating interactions between folded components and highly charged, very long and flexible protein segments. Our work offers the first glimpse of a molecular model for knobs.
References:
- Oberli A, Zurbrügg L, Rusch S, Brand F, Butler ME, Day JL, Cutts EE, Lavstsen T, Vakonakis I, Beck HP (2016) Plasmodium falciparum PHIST Proteins Contribute to Cytoadherence and Anchor PfEMP1 to the Host Cell Cytoskeleton. Cell Microbiol. 18, 1415-28.
- Warncke JD, Vakonakis I, Beck HP (2016) PHIST proteins, at the center of host cell remodeling
- Watermeyer JM, Hale VL, Hackett F, Clare DK, Cutts EE, Vakonakis I, Fleck RA, Blackman MJ, Saibil HR (2016) A spiral scaffold underlies cytoadherent knobs in Plasmodium falciparum-infected erythrocytes. Blood. 127, 343-51.
- Oberli A, Slater LM, Cutts E, Brand F, Mundwiler-Pachlatko E, Rusch S, Masik MFG, Erat MC, Beck HP, Vakonakis I (2014) A Plasmodium falciparum PHIST protein binds the virulence factor PfEMP1 and co- migrates to knobs on the host cell surface. FASEB J. 28, 4420-33.
- Boddey JA, Cowman AF (2013) Plasmodium nesting: remaking the erythrocyte from the inside out. Annu Rev Microbiol. 67, 243-69.
David F. Sargent
ETH Zurich, Switzerland
Title: Microrobotics enables non-contact, fully automated protein crystal harvesting

Biography:
David Sargent obtained his PhD in biophysics from the University of Western Ontario, Canada, followed by postdoctoral studies at the ETH Zurich and the University of Sydney (Australia). He has had extensive experience in macromolecular crystallography at the ETH Zurich, and recently has also been associated with the Multiscale Robotics Laboratory (ETH Zurich) of Bradley J. Nelson. David is one of the founders of MagnebotiX, a spinoff of the ETH, which provides tools for magnetic propulsion and guidance at the microscopic scale. The work reported here uses this technology to streamline and accelerate the process of macromolecular crystal structure determination.
Abstract:
Statement of the Problem: Most aspects of macromolecular structure determination, from synthesis and purification of materials, through crystallization, data collection and model building, are highly automated, but the recognition, harvesting and cryocooling of crystals remains a predominantly manual task. Several concepts, including in situ crystallography, are being developed to overcome these difficulties, but frequently impose other restrictions, such as on data collection strategies. We are developing hardware and software to support crystal harvesting using standard crystallization procedures, thus avoiding such limitations. Methodology & Theoretical Orientation: We use a magnetically driven, mobile, rolling microrobot, the “RodBot”, to locally move the liquid surrounding a crystal, and the crystal then passively follows the flow. Crystal position is monitored using low level uv-light. Transport is controlled using flexible algorithms that allow for error-recovery following stochastic disturbances. Findings: We demonstrate the effectiveness of the technique using crystals of different geometries and densities in a variety of buffers and cryoprotectants. Even at this developmental stage average harvesting time is reduced compared to manual operations. Conclusion & Significance: This non-destructive, non-contact method allows crystals to be extracted reliably from the growth droplet in a completely automated process. Harvesting can take place remotely in climate-controlled chambers, ensuring optimal conditions throughout the process with respect to temperature, humidity and composition of the environment. Damage to valuable crystals due to operator jitter or fatigue is eliminated. Incorporation into existing robotics setups for sample handling will also allow increased reproducibility of flash-cooling. Fully automated structure determination pipelines using well-established techniques are now possible and will yield improved data quality at reduced cost.
References:
- Zeydan B, Petruska AJ, Somm L, Pieters RS, Fang Y, Sargent DF, Nelson BJ (submitted) Automated protein crystal harvesting.
- Pieters RS, Lombriser S, Alvarez-Aguirre A, Nelson BJ (2016) Model predictive control of a magnetically guided rolling microrobot. IEEE Robotics and Automation Letters 1.1: 455-460.
- Charreyron S, Pieters RS, Tung HW, Gonzenbach M, Nelson BJ (2015) Navigation of a rolling microrobot in cluttered environments for automated crystal harvesting. Paper at IEEE/RSJ Int. Conf. Intell. Robots and Systems (IROS), Germany 2015
- Tung HW, Sargent DF, Nelson BJ (2014) Protein crystal harvesting using the RodBot: a wireless mobile microrobot. J Appl. Cryst. 47: 692-700.
- Tung HW, Peyer K, Sargent DF, Nelson BJ (2013) Noncontact manipulation using a transversely magnetized rolling robot. Appl. Phys. Letters 103.11:114101.
Toshiya Senda
High Energy Accelerator Research Organization (KEK), Japan
Title: The native (Sulfur) SAD method in Photon Factory
Biography:
Toshiya Senda has completed his PhD from Nagaoka University of Technology (Niigata, Japan) in 1995. He was a research associate in Nagaoka University of Technology (1995-2001) and a senior researcher in Institute of Advanced Industrial Science and Technology (2001-2012). Now, he is the director/professor of Structural Biology Research Center of High Energy Accelerator Research Organization (KEKI) in JAPAN. He was awarded the CrSJ (Crystallographic society of Japan) award in 2014 (Structural biology studies of CagA from Helicobacter pylori and histone chaperon CIA/ASF1).
Abstract:
Crystallography has been a major method to determine 3D structures of biological macromolecules at atomic resolution. While a new method with cryo-EM is becoming another major technique for the 3D structure analysis, crystallography still has some advantages. Recently, many crystal structures of biological macromolecules are determined by MAD/SAD method with seleno-methionine proteins (SeMet-proteins). Since selenium has an X-ray absorption edge near 1Å, it is convenient to utilize in the MAD/SAD method. While this technique is useful, crystallographers need to prepare SeMet-proteins. If we can develop a method to solve the phase problem without using SeMet-proteins, it would be highly useful for crystallographers. So, we have tried to develop the native SAD (or sulfur SAD) method, in which anomalous signals from sulfur atoms in the native protein are utilized. However, there are some problems in the native SAD method. First, sulfur gives only weak anomalous signals with X-ray typically utilized in protein crystallography (X-ray wavelength of around 1Å). To increase the anomalous signals, we need to use a longer wavelength X-ray than usual. However, since a longer wavelength X-ray shows significant absorption by air, solvent, protein etc., data quality is degraded by the absorption. The native SAD method, therefore, requires a specific system for high quality data collection. To achieve routine utilization of the native SAD method, we have developed a beamline (BL-1A) dedicated for the native SAD method. In BL-1A, we can utilize a long wavelength X-ray (1.9 – 3.5 Å). Furthermore, the goniometer and X-ray detector are installed inside a He chamber to prevent the absorption problem. Our system enables us to solve crystal structures of proteins by the native SAD method. In the presentation, we will present several examples of crystal structure determination with native SAD. Also, we will mention our unique method for crystal freezing to collect high quality diffraction data required in native SAD experiments.
References:
- Liebschner, D., Yamada, Y., Matsugaki, N., Senda, M. & Senda, T. (2016). On the influence of crystal size and wavelength on native SAD phasing. Acta Crystallogr. D72, 728–741.
- Hiraki, M., Matsugaki, N, Yamada, Y. & Senda, T. (2016) Development of sample exchange robot PAM-HC for beamline BL-1A at the photon factory. API Conf. Proc. 1741, 030029.
- Nagae, M., Liebschner, D., Yamada, Y., Morita-Matsumoto, K., Matsugaki, N., Senda, T., Fujita, M., Kinoshita, T., Yamaguchi, Y. (2017) Crystallographic analysis of murine p24g2 Golgi dynamics domain. Proteins, 85, 764-770. doi: 10.1002/prot.25242
- Nagae, M., Mishra, S. K., Neyazaki, M., Oi, R., Ikeda, A., Matsugaki, N., Akashi, S., Manya, H., Mizuno, M., Yagi, H., Kato, K., Senda T., Endo, T., Nogi, T. & Yamaguchi Y. (2017) Genes to Cells, doi 10.1111/gtc.12480 [Epub ahead of print]
- Senda, M., Hayashi, T., Hatakeyama, M., Takeuchi, K., Sasaki, A. T. & Senda, T. (2016) Use of multiple cryoprotectants to improve diffraction quality from protein crystals. Crystal Growth & Design 16, 1565-1571. Doi: 10.1021/acs.cgd.5b01692
Yawen Bai
NIH, USA
Title: Structural mechanisms of nucleosome recognition as revealed by methyl-TROSY
Time :

Biography:
Yawen Bai received his Ph.D. in Biophysics from the University of Pennsylvania Medical School. After postdoctoral work at the Scripps Research Institute, La Jolla, California, he became an investigator at the National Cancer Institute of the National Institutes of Health in Bethesda, Maryland since 1997. The research interests of his group include structural studies on protein folding intermediates, histone chaperones, epigenetic specification of centromeres and chromatin folding.
Abstract:
Human genome is packaged into chromatin through association with small positively charged histone proteins. The structural unit of chromatin is the nucleosome, which consists of ~147 bp of DNA and two copies of each of the four core histones (H2A, H2B, H3 and H4). Numerous proteins regulate chromatin structure and function through specific binding to the nucleosome. The structural basis of many of these interactions is unknown. Structural determination of the nucleosome in complex with a protein by X-ray crystallography and single particle cryo-EM has proven to be very challenging in many cases due to difficulties to crystalize them and dissociation of the complex during cryo processes. On the other hand, the nucleosome is too large (> 200 KDa) for structural studies with conventional NMR methods. We have used methyl-TROSY [1] coupled with site-specific mutagenesis and paramagnetic spin labeling to investigate how the nucleosome is recognized by various chromatin factor proteins, including high-mobility group nucleosomal protein [2], centromere protein C [3] and linker histones [4,5]. Major results and future perspectives will be presented.
References:
- Tugarinov V, Hwang JE, Ollerenshaw JE, Kay LE. (2003) Cross-correlated relaxation enhanced 1H-13C NMR spectroscopy of methyl groups in very high molecular weight proteins and protein complexes. J Am Chem Soc 125, 10420-10428.
- Kato H, van Ingen H, Zhou BR, Feng H, Bustin M, Kay LE, Bai Y. (2011) Architecture of the high mobility groups nucleosomal protein 2-nucleosome complex as revealed by methyl-based NMR. Proc Natl Acad Sci 108, 12283-12288.
- Kato H, Jiang J, Zhou BR, Rozendaal M, Feng H, Ghirlando R, Xiao TS, Straight AF, Bai Y. (2013) A conserved mechanism for centromeric nucleosome recognition by centromere protein CENP-C. Science, 340, 1110-1113.
- Zhou BR, Feng H, Kato H, Dai L, Yang Y, Zhou Y, Bai Y. (2013) Structural insights into the histone H1-nucleosome complex. Proc Natl Acad Sci 110, 19390-19395.
- Zhou BR, Jiang J, Feng H, Ghirlando R, Xiao TS, Bai Y. (2015) Structural mechanisms of nucleosome recognition by linker histones. Mol Cell 58, 628-638.
Biography:
Julien Boudet received his PhD degree in structural biology and biophysics from the University of Grenoble (Joseph Fourier University) in France under the supervision of Prof. Jean-Pierre Simorre. During his thesis, he learned nuclear magnetic resonance (NMR) spectroscopy and used this powerful method to investigate proteins and oligonucleotides structures, molecular mechanisms underlying antibiotic resistance and viral proteins interactions. After graduating, Julien joined the group of Prof. Frédéric Allain in ETH Zurich as a researcher. He focused his investigations on the DNA replication machinery and, in particular on the primase-mediated catalysis. He set up innovative computational methods to investigate challenging biological systems and demonstrated the role of cofactors in improving the pRN1 primase specific template recognition. His currently developing computational and analysis tools for structural biology.
Abstract:
Primases are single-stranded DNA dependent polymerases that synthesize RNA/DNA primers during replication. A primase, a DNA polymerase and an helicase compose the replication machinery of the archaeal plasmid pRN11. The structure of the archaeal functional primase domain has been solved by X-ray crystallography 2,3 and it revealed an heteromeric structure with a catalytic prim/pol domain tethered to a novel helix bundle domain.
We investigated the NMR structure of the functional pRN1 primase domain in complex with a single-stranded DNA template containing the GTG motif 4. We showed that the catalytic prim/pol domain of this 38 kDa enzyme is not required for template binding. Intermolecular contacts detected exclusively between the helix bundle domain and the DNA led us to isolate specifically this structurally independent unit. Our results are compatible with a conformational switch between a template-bound open state and a closed active complex 3,5,6.
We solved the solution structures of the helix bundle domain in complex with the single-stranded DNA template alone and upon cofactors addition. Affinity measurements validated our structural data demonstrating the importance of residues located in helices 10 and 12 for the interaction with the GTG motif and confirmed the specificity improvement observed upon cofactors binding.
In association with functional assays, these novel transient structures bring new perspectives and will help us to characterize the molecular steps required for priming.
References:
- Lipps G., Röther S., Hart C. and Krauss G. (2003) EMBO J, 22, 2516-25.
- Lipps G., Weinzierl AO., von Scheven G., Buchen C. and Cramer P. (2004) Nat. Struct. Mol. Biol., 11, 157-62.
- Beck K., Vannini A., Cramer P. and Lipps G. (2010) Nucleic Acids Res., 38, 6707-18.
- Beck K. and Lipps G. (2007) Nucleic Acids Res., 35, 5635-45.
- Lipps G. (2011) Biochem Soc Trans., 39, 104-6
Luis Paulo. B. Scott
Universidade Federal do ABC , Brazil
Title: A new protocol to investigate conformational population patterns in the enzymatic activity cycle of proteins using molecular dynamics and normal mode analysis

Biography:
Luis Paulo Barbour Scott is Associate Professor in Federal University of UAFBC. He has his expertise in conformational changes and functional movements of macromolecules, specially proteins. Over the last four years, Dr. Luis Paulo Scott’s research group has been financed to investigate molecules related to neurodegeneration and aggregate formation by means of normal mode analysis and molecular dynamics combined. The laboratory coordinated by Dr. Luis Paulo Scott has become more and more specialized in the study of macromolecules structural dynamics (functional movements in collaboration with Dr. David Perahia from France.
Abstract:
Human immunodeficiency virus type-1 protease (HIV-1 PR) is an aspartic protease whose proteolytic activity is essential for cleaving precursor viral polyproteins into individual proteins implied in viral replication. Once HIV enters within a host cell, its RNA is transcribed into DNA through reverse transcriptase, integrated and amplified along with the replication of the host cell's DNA. Gag and gag-pro-pol genes are transcribed into messenger RNA, translated into gag and gag-pro-pol precursors proteins in the cytoplasm, and then assembled at the cell surface for budding and formation of the immature viral particles. In this work, we propose a computational protocol to generate and select HIV protease conformations relevant to its function using Normal Mode Analysis (NMA). We have considered structures of the apoenzyme, the protein with its substrate and product and the protein with a drug. This set of structures should reveal large amplitude motions that are critical to the protease activity cycle as: i) substrate acquisition; ii) substrate cleavage and; iii) product release. The apoenzyme presents an increased flap conformational diversity compared to the various complexes, predominantly populated with open flap conformations, that can possibly be related to the substrates acquisition. The enzyme-substrate complexes show more structural diversity than enzyme-product complexes, suggesting a role of these conformational changes in catalytic activity. We presents a promising protocol to identify the conformational diversity induced by different types of ligands and that can help the drug design process.
References:
- Lima, A N. ; Philot, E. A. ; TROSSINI, G. ; SCOTT, L. P. B. ; MALTAROLLO, V. ; HONORIO, K. M. . Use of machine learning approaches for novel drug discovery. Expert Opinion on Drug Discovery (Print), v. 11, p. 225-239, 2016.
- MEISSNER, G. ; RESENDE-LARA, P. ; MATSUBARA, F. ; Luis P B Scott ; BRAZ, ANTÔNIO S.K. ; CHAVES-MOREIRA, D. ; SOARES, E. ; TREVISAN-SILVA, D. ; GREMSKI, L. ; VEIGA ; CHAIM, O. . Molecular cloning and in silico characterization of knottin peptide, U2-SCRTX-Lit2, from brown spider (Loxosceles intermedia) venom glands. Journal of Molecular Modeling (Online), v. 22, p. 196, 2016.
- ARAUJO, G. ; Silva, R H T ; SCOTT, L. P. B. ; ARAUJO, A. S. ; SOUZA, F. P. ; OLIVEIRA, R. J. . Structure and functional dynamics characterization of the ion channel of the human respiratory syncytial virus (hRSV) small hydrophobic protein (SH) transmembrane domain by combining molecular dynamics with excited normal modes. Journal of Molecular Modeling (Print) , v. 22, p. 3-8, 2016.
- Philot, E. A. ; LOPES, D. M. ; SOUZA, A. T. ; BRAZ, A. S. K. ; NANTES, I. L. ; RODRIGUES, T. ; Perahia, D. ; MITEVA, M. A. ; SCOTT, L. P. B. . Binding of phenothiazines into allosteric hydrophobic pocket of human thioredoxin 1. European Biophysics Journal, v. 43, p. 2798-286, 2016.
- CONTO, V. ; BRAZ, A. S. K. ; Perahia, D. ; SCOTT, L. P. B. . Recovery of the wild type atomic flexibility in the HIV-1 protease double mutants. Journal of Molecular Graphics & Modelling , v. 59c, p. 107-116, 2015.
Thomas Braun
University of Basel, Switzerland
Title: Cryo-Electron Microscopy Grid Preparation from Nanoliter-Sized Protein Samples and Single-Cell Extracts

Biography:
Thomas Braun received his Ph.D. 2002 in biophysics from the Biozentrum, University of Basel, Switzerland. During his Ph.D. thesis he applied high-resolution electron microscopy and digital image processing to study the structure and function of membrane proteins. Subsequently, he worked on nanomechanical sensors to characterize the mechanics of membrane proteins at the Institute of Physics, University Basel and the CRANN, Trinity College Dublin, Ireland. He has been working at the Center for Cellular Imaging an NanoAnalytics (Biozentrum, University of Basel, Switzerland) since 2009 and is developing new methods for electron microscopy, single cell analysis and nanomechanical sensors for biological applications.
Abstract:
Cryo-electron microscopy (cryo-EM) sample preparation techniques ensure that biological specimens can be investigated at physiological conditions in the electron microscope. However, these preparation methods suffer from extensive blotting steps leading to a massive loss of sample and sometimes to partial denaturation of sensitive protein complexes. We have developed a simple method for the almost lossless conditioning and preparation of nanoliter-volumes of biological samples for EM. The method does not involve any blotting steps. A microcapillary is used to aspirate 3 to 20 nanoliters of sample, depending on the experiment (Figure 1A). The sample is applied (left) and spread (center) on the EM-grid. Real-time monitoring allows the thickness of the water film to be assessed and decreased to the optimum value prior to vitrification (right). We prepared cryo-EM grids of various samples, e.g., bacteriophages and soluble proteins as shown in Figure 1B&C, to demonstrate the usefulness and general applicability of the method. We also showed that high-resolution 3D structures can be calculated from single-particle preparations of a soluble protein. In addition to cryo-EM grid preparation, the versatile method allows nanoliter-sized sample volumes to be conditioned for EM, e.g., negatively stained with heavy metal salts or embedded in trehalose.
In addition, we combine the new sample preparation method with a single cell lysis device for adherent eukaryotic cells and image the aspirated cell contents by TEM. To demonstrate the usefulness of this new “visual proteomics” approach we visualized the changes occurring in single cell proteomes upon heat shocking the cells. Furthermore, we have developed a protein-fishing method based on a magnetic trap and photo-cleavable composite material, to ‘fish’ untagged proteins from cell lysate by antibodies. This allows target proteins to be isolated from approx. 40’000 cells in 90 min and analysed by EM.
References:
- Kemmerling S, Ziegler J, Schweighauser G, Arnold S A, Giss D, Müller S A, Ringler P, Goldie K N, Goedecke N, Hierlemann A, Stahlberg A, Engel A, Braun T (2012) Connecting µ-fluidics to electron microscopy. Journal of Structural Biology, 177:1, 128–134.
- Arnold S A, Albiez S, Opara N, Chami M, Schmidli C, Bieri A, Padeste C, Stahlberg H, Braun T (2016) Total sample conditioning and preparation of nanoliter volumes for electron microscopy. ACS Nano, 10: 5, 4981–4988.
- Arnold S A, Albiez S, Bieri A, Syntychaki, A, Adaixo R, McLeod R A, Goldie K N, Stahlberg H, Braun T. (2016) Blotting-free and lossless cryo-electron microscopy grid preparation from nanoliter-sized protein samples and single-cell extracts. Journal of Structural Biology, in press.
- Kemmerling S, Arnold S A , Bircher B, Sauter N, Escobedo C, Dernick G, Hierlemann A, Stahlberg H, Braun T (2013) Single-cell lysis for visual analysis by electron microscopy. Journal of Structural Biology, 183: 3, 467–473.
- Giss D, Kemmerling S, Dandey V, Stahlberg H, Braun T (2014) Exploring the interactome: microfluidic isolation of proteins and interacting partners for quantitative analysis by electron microscopy. Analytical Chemistry, 86: 10, 4680–4687.
- Track 2: Molecular Modeling and Drug Designing
Location:
Session Introduction
Michael Hennig
leadXpro AG, Villigen, Switzerland
Title: Structure Based Drug Discovery on Membrane Protein Targets: New developments and advancements

Biography:
Michael Hennig is a drug discovery research manager with 22 years of experience in pharmaceutical industry. He co-founded and is CEO and Chairman of the board of leadXpro, an emerging biotech company and spin-out of the Paul Scherrer Institute (ETH, Switzerland) that is dedicated to structure based drug discovery of membrane protein targets. Formerly he worked 20 years at Roche research Basel, Global Head and Principle Leader of discovery technologies with responsibility for structure based drug discovery, protein science, assay development and HTS, corporate compound library, stem cell platform. In addition, he is guest Professor at the University of Basel in structural biology, gives lecture series in pharmacy, is author of >75 paper and lecturer at conferences, inventor of 8 patents in areas of technology, discovery and formulation of drug substances.
Abstract:
Today, structure based drug discovery is well implemented in the drug discovery engine of many pharmaceutical companies. Whereas soluble proteins are managed well within the project timelines and portfolio changes in pharmaceutical industry, transmembrane proteins still represent a significant challenge. LeadXpro combines expertise of drug discovery, excellence in high quality solubilized and purified membrane protein science and use of cutting edge biophysical methods like X-ray data collection at synchrotron and FEL sources, single particle cryo-electron microscopy, SPR and others. Strong relationship between leadXpro and Swiss large research facilities like PSI-SLS and SwissFEL as well as C-CINA will enable advances in structure determination of challenging membrane protein drug targets that have not been feasible before. Knowledge of the drug candidate and protein target 3D-structure, together with the full characterization of its interaction by biophysical binding and functional assays will enable to generate novel and better lead molecules for future medicines.
Examples of recent developments include the successful fragment screening for the GPCR neutrotensin receptor 1, a fragment screening with 6369 compounds was performed with SPR and 44 hits identified. Finally, 4 selected hits were validated in NMR experiments and computational analysis gave insight into the potential fragment-binding location and interactions to inspire further chemistry efforts. Furthermore, serial crystallography performed at synchrotron and free electron laser enables structure determination on challenging drug targets. Advantages are analysis at physiologically more relevant room temperature (no freezing of crystals required), low or no radiation damage and the use of very small crystals.
References:
- Renaud, J-P., Chung, C-W., Danielson, U.H., Egner, U., Hennig, M., Hubbard, R.E., Nar, H. Biophysics in drug discovery : impact, challenges and opportunities. Nature Reviews Drug Discovery 15, 679-698 (2016)..
- Huber, S., Casagrande, F., Hug, M.N., Wang, L., Heine, P., Kummer, L., Plückthun, A., Hennig, M. SPR-based fragment screening with neurotensin 1 generates novel small molecule ligands. PlosONE, 2017
- Bocquet, N., Kohler, J; Hug, M.N., Kusznir, E., Rufer, A.C.,Dawson, R.J., Hennig, M., Ruf, A, Huber, W., Huber, S., Real time monitoring of binding events on a thermostabilized human A2A receptor embedded in a lipid bilayer by surface plasmon resonance. BBA- Biomembranes, 2015, Biochim. Biophys Acta 1848 (5), 1224-1233 (2015).

Biography:
Dirksen Bussiere Led 22-FTE structural biology and biophysics research effort (X-ray crystallography, biophysics, NMR and protein biochemistry support) on multiple structure-based projects encompassing variety of anti-infective, metabolic, and oncology targets
Led fragment-based screening and biophysics team which spans four functional areas (Structural and Biophysical Chemistry, Computational Chemistry, Biochemical Lead Discovery, and Protein Sciences)
Led multiple hit-finding project teams focused on finding hits for anti-infective and oncology
targets
Served as lead structural biologist. biochemist and biophysicist on multiple oncology and anti-infective drug discovery project teams
Provided molecular modeling, drug design, and bioinformatics support to project teams
Provided fragment-based screening support for project teams, including NMR and biophysical screening
Named Novartis Leading Scientist in 2007, an award & title given to less than 1% of Novartis scientists world-wide
Abstract:
Several biological functions, particularly chromosome segregation, require the generation of motile force. The generation of this force relies heavily on a class of proteins known as motor proteins. Motor proteins such as Kinesin Spindle Protein (KSP), also known as Eg5, utilize the energy derived from ATP hydrolysis to generate motile force. High-throughput screening of Eg5 identified several hits which were non-competitive with ATP with micromolar IC50’s capable of inhibiting the motor protein. Using structure-based drug design, these hits were progressed to NVP-BQS481, a clinical candidate with an IC50 of 700 picomolar. The talk will present the design concepts and optimization techniques used to advance the series to the pre-clinical stage.
References:
- Barsanti, PA, Wang, W, Duhl, D, Brameier, N, Martin, E, Bussiere, D, Walter AO (2010) The discovery of tetrahydro-beta-carbolines as inhibitors of the kinesin Eg5. Bioorg Med Chem Lett 20(1): 157-160.
- Murray, JM, Bussiere DE (2009) Targeting the purinome. Methods Mol Biol 575: 47-92.
- Mayer, TU, Kapoor, TM, Haggarty, SJ, King RW, Schreiber, SL, Mitchison, TJ (1999) Small molecule inhibitor of mitotic spindle bipolarity identified in phenotype-based screen. Science 286: 971-974.
- Duhl, DM, Renhowe PA (2005) Inhibitors of kinesin motor proteins—research and clinical progress. Curr Opin Drug Discov Devel 8(4): 431-436.
- Bergnes, G, Brejc, K, Belmont, L (2005) Mitotic kinesins: prospects for antimitotic drug discovery. Curr Top Med Chem 5(2): 127-145.
Christina Scharnagl
Technical University of Munich, Germany
Title: Does the dynamics of their transmembrane domain qualify bitopic membrane proteins as substrates for intramembrane proteolysis?

Biography:
Christina Scharnagl has her expertise in molecular dynamic simulations of membrane proteins. Her work focuses on biophysical principles of the interdependence of transmembrane helix dynamics, helix-helix binding, and helix-lipid interactions. In silico modelling and advanced computational analysis are closely connected to experimental work in research collaborations in order to interpret and guide the experiments and to validate the simulations. The aim of the joint efforts is to understand the impact of these phenomena on multiple biological processes, such as membrane fusion and intramembrane proteolysis.
Abstract:
Integral membrane proteins facilitate communication between the inside of the cell and its exterior. Their transmembrane domains (TMDs) support a diversity of biological functions and exhibit sequence-dependent conformational dynamics on multiple size and time scales. Membrane proteins are notoriously difficult to study by experimental methods. Molecular dynamics (MD) simulations provide a powerful tool of high spatial and temporal resolution that effectively complements experimental methods. Here we focus on the conformational dynamics of the TMD of the amyloid precursor protein (APP). APP is enzymatically hydrolyzed within its TMD by g-secretase (GSEC), forming toxic Ab peptides regarded as molecular cause of Alzheimer's disease (AD). Besides APP, GSEC cleaves ~100 single-span membrane proteins within their TMDs, however without obvious consensus sequence. Finding the link between the molecular architecture of the substrate TMDs and cleavage is, therefore, of upmost importance. Because unfolding is obvious to expose the scissile bond, it seems plausible that the TMD itself is optimized for local helix unwinding. However, this view was challenged by our experiments and MD simulations. Our results suggest an alternative model where reaching a cleavage-competent state involves multiple conformational transitions of the substrate/enzyme complex where global conformational plasticity of the substrate TMD is a key determinant. In a first step, we compare the conformational flexibility of a large number of substrate and non-substrate TMDs, as well as TMDs carrying missense mutations related to early onset AD. Knowing the key-dynamical motifs will help to identify new substrates and to elucidate the physiological functions of the protease in the brain and other organs. This work is part of a collaborative research program (https://www.i-proteolysis.de/).
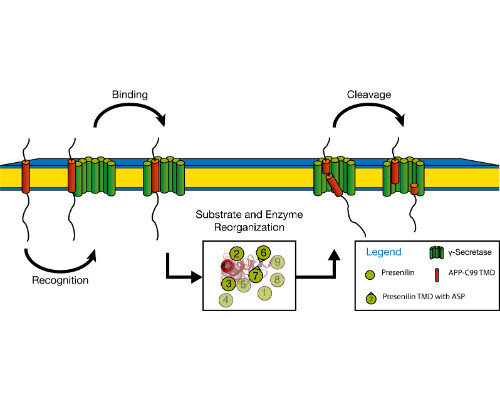
References:
- Langosch C, Scharnagl C, Steiner H, Lemberg M (2015) Understanding intramembrane proteolysis: from protein dynamics to reaction kinetics. Trends Biochem. Sci. 40:318-327.
- Scharnagl C, Pester O, Hornburg P, Hornburg D, Götz A, Langosch D (2014) Side-chain to main-chain hydrogen bonding controls intrinsic backbone dynamics of the amyloid precursor protein transmembrane helix. Biophys. J. 106: 1318-1329.
- Pester O, Götz A, Multhaup G, Scharnagl C, Langosch D (2013) The cleavage domain of the amyloid precursor protein transmembrane helix does not exhibit above-average backbone dynamics. ChemBioChem 14:1943-1948.
- Pester O, Barrett P, Hornburg D, Hornburg P, Pröbstle R, Widmaier S, Kutzner C, Dürrbaum M, Kapurniotu A, Sanders CR, Scharnagl C, Langosch D (2013) The backbone dynamics of the amyloid precursor protein transmembrane helix provides a rationale for the sequential cleavage mechanism of g-secretase. J. Am. Chem. Soc. 135: 1317-1329.
- Ried C, Scharnagl C, Langosch D (2015) Entrapment of water at the transmembrane helix-helix interface of quiescin sulfhydryl oxidase 2. Biochemistry 55: 1287-1290.
Andreas Kuhn
University of Hohenheim, Germany
Title: The dynamics of a protein during its insertion into a membrane

Biography:
Andreas Kuhn has his expertise in protein folding of membrane proteins. Studies include reconstituted systems with bacterial translocases and insertase, as well in vivo studies with Escherichia coli. For biophysical experiments the membrane proteins are purified and their folding is monitored spectroscopically in real time after their addition to liposomes. Andreas Kuhn obtained his PhD from the Universities Basel and Freiburg i. Br. 1982. After a postdoc at UCLA with Bill Wickner he continued at the Biozentrum Basel from 1986 to 1989 and accepted a professorship at the University Karlsruhe. Since 1996 he is at the University of Hohenheim in Stuttgart.
Abstract:
Most membrane proteins are inserted co-translationally by the Sec-translocase or the YidC/Oxa1/Alb3 insertases. The folding of these proteins occurs within the membrane during the interaction with the insertases. We have purified and reconstituted YidC, the membrane insertase of Escherichia coli. The protein spans the membrane 6 times, and the recently solved structure shows a hydrophilic cavity and a greasy slide between the transmembrane segments TM3 and TM5. Hydrophobic residues of TM3 and TM5 interact with the substrate, in particular with a prospective transmembrane segment of an inserting membrane protein as we documented by disulfide crossinking experiments.
The membrane insertion process can be studied with the reconstituted vesicle system. The purified substrate proteins are solubilized in 10% isopranol or kept unfolded with urea or GuHCl. When the substrate proteins are added to the proteoliposomes by dilution 1:100, they rapidly bind to YidC and become membrane inserted within 2 msec. FRET-based kinetic measurements show that the substrate proteins approach YidC to a close distance during the insertion event. Time-resolved fluorenscence anisotropy shows that the periplasmic domain of YidC moves when a substrate protein was added. This suggests that both the insertase and the substrate protein undergo conformational motions.
References:
- Winterfeld, S., S. Ernst, M. Börsch, U. Gerken and A. Kuhn (2013). Real time observation of single membrane protein insertion events by the E. coli insertase YidC. PLOSone 8, e59023.
- Dalbey, R.E. and A. Kuhn A. (2014) How YidC inserts and folds proteins across a membrane. Nature Struc.& Mol.Biol. 21, 435-436.
- Dalbey, R. E. and A. Kuhn (2015). Membrane insertases are present in all three domains of life. Structure 23, 1559-1560.
- Proß, E., L. Soussoula, I. Seitl, D. Lupo and A. Kuhn (2016). Membrane targeting and insertion of the C-tail protein SciP. J. Mol. Biol. 428, 4218-4227.
- Kuhn, A. and R.E. Dalbey (2016). A conformational crosstalk between SecA and SecY opens the SecYEG channel. Current Biol. 26, 811-813.
Shuanghong Huo
Clark University, USA
Title: Extract the thermodynamic and kinetic information from protein simulations using dimensionality reduction

Biography:
Shuanghong Huo received her Ph.D. in Computational Chemistry from Boston University. She had postdoctoral training at UC-San Francisco. She is a Professor of Chemistry and Biochemistry at Clark University, Worcester, USA. Her research interest is protein folding, misfolding, and aggregation. Recently her group is developing dimensionality reduction methods and graph representations of protein free energy landscapes.
Abstract:
In the study of protein thermodynamics and kinetics it is of paramount importance to characterize protein free energy landscapes. Dimensionality reduction is a valuable tool to complete the task. We have evaluated several methods of dimensionality reduction, including linear and non-linear methods, such as principal component analysis, Isomap, locally linear embedding, and diffusion maps. A series of criteria was used to assess different aspects of the embedding qualities. Our results have shown that there is no clear winner in all aspects of the evaluation and each dimensionality-reduction method has its limitations in a certain aspect. The linear method, principal component analysis, is not worse than the nonlinear ones in some aspects for our peptide system. We have also developed a mathematical formulation to demonstrate that an explicit Euclidean-based representation of protein conformation space and the local distance metric associated to it improve the quality of dimensionality reduction. For a certain sense, clustering protein conformations into macro-clusters to build a Markov state model is also an approach of dimensionality. We have tested inherent structure and geometric metric for state space discretization and demonstrated that the macro-clusters based on inherent structure give a meaningful state space discretization in terms of conformational features and kinetics.
References:
- H. Liu, M. Li, J. Fan and S. Huo (2016) Inherent Structure Versus Geometric Metric for State Space Discretization. J. Compt. Chem. 37:1252-1258.
- M. Duan, H. Liu, M. Li, L. Han and S. Huo (2015) Network representation of conformational transitions between hidden intermediates of Rd-apocytochrome b562. J. Chem. Phys. 143: 135101-10.
- M. Duan, M. Li, L. Han, and S. Huo (2014) Euclidean sections of protein conformation space and their implications in dimensionality reduction. Proteins: Structure, Function, and Bioinformatics. 82: 2585-2596.
- M. Li, M. Duan, J. Fan, L. Han and S. Huo (2013) Graph representation of protein free energy landscape. J. Chem. Phys. 139: 185101-8.
- M. Duan, J. Fan, M. Li, L. Han and S. Huo (2013) Evaluation of Dimensionality-Reduction Methods from Peptide Folding–Unfolding Simulations. J. Chem. Theory Comput. 9: 2490-2497.
Marek Cieplak
Institute of Physics PAS, Poland
Title: Dynamics of knotted and entangled neurotoxic polypeptides

Biography:
Head, Laboratory of Biological Physics, Institute of Physics, Polish Academy of Sciences in Warsaw, Poland. Education - M.S., Department of Physics, University of Warsaw, 1973; Ph. D., Department of Physics, University of Pittsburgh, 1977; D.Sc., Department of Physics, University of Warsaw, 1984. Professorial title, 1994. Fields of interest – condensed matter theory (spin waves, spin glasses, porous media, growth processes, atomic friction, river networks, nanofluidics, self-organized nanostructures) and biological physics (large conformational changes of biomolecules within coarse-grained models, especially as induced by stretching, proteins with knots and slipknots, protein folding, dynamics of virus capsids and other multi-proteinic structures such as a cellulosome, interaction of proteins with solids, proteins at air-water interface, modeling of proteasomes, inference of genetic networks from the microarray data). Co-author of textbook “Theory of Quanta”, Oxford University Press 1992. 250 research papers.
Abstract:
We review the physics of processes involving large conformational transformations in knotted proteins in bulk water and then consider folding in ribosomes and unfolding in proteasomes. Formation of a knot is demonstrated to be facilitated by the nascent conditions at the ribosome. Knots in proteins have been proposed to resist proteasomal degradation. Ample evidence associates proteasomal degradation with neurodegeneration. One interesting possibility is that indeed knotted conformers stall this machinery leading to toxicity. However, although the proteasome is known to unfold mechanically its substrates, at present there are no experimental methods to emulate this particular traction geometry. Here, we consider several dynamical models of the proteasome in which the complex is represented by an effective potential with an added pulling force. This force is meant to induce translocation of a protein or a polypeptide into the catalytic chamber. The force is either constant or applied periodically. The translocated proteins are modelled in a coarse-grained fashion. We do comparative analysis of several knotted globular proteins and the transiently knotted polyglutamine tracts of length 60 alone and fused in exon 1 of the huntingtin protein. Huntingtin is associated with Huntington disease, a well-known genetically-determined neurodegenerative disease. We show that the presence of a knot hinders and sometimes even jams translocation. We demonstrate that the probability to do so depends on the protein, the model of the proteasome, the magnitude of the pulling force, and the choice of the pulled terminus. In any case, the net effect would be a hindrance in the proteasomal degradation process in the cell. This would then yield toxicity via two different mechanisms: one through toxic monomers compromising degradation and another by the formation of toxic oligomers.
References:
- Zhao Y, Chwastyk M, Cieplak M (2017) Topological transformations in proteins: effects of heating and proximity of an interface. Sci. Rep. 7:39851alcohol-related braindamage. Alcohol Alcohol 44:136-140.
- Wojciechowski M, Cieplak M (2016) Dual binding mode in cohesin-dockerin complexes as assessed through stretching. J. Chem. Phys. 145:134102.
- Wolek K, Cieplak M (2016) Criteria for folding in structure-based models of proteins. J. Chem. Phys. 144:185102.
- Wojciechowski M, Gomez-Sicilia A, Carrion-Vazquez M, Cieplak M (2016) Unfolding knots by proteasome-like systems: simulations of the behavior of folded and neurotoxic proteins. Mol. Biosyst. 12:2700-2712.
- Gomez-Sicilia A, Sikora M, Cieplak M, Carrion-Vazquez M (2015) An exploration of the universe of polyglutamine structures. PLOS Comp. Biol. 11:e1004541.

Biography:
Chantal Prévost is researcher at the Theoretical Biochemistry Laboratory (LBT) of the French National Research Center (CNRS), in Paris. She has developed a large expertise in studying macromolecular self-assembliy in silico, either by elaborating new algorithms for flexible proteins docking or by studying fundamental biological processes involving the transition between instable conformational substates. She presently applies this expertise to exploring the architecture or oligomeric assemblies as well as elucidating the mechanism of homologous recombination, in collaboration with experimental partners.
Abstract:
Statement of the Problem: Many proteins present highly flexible or disordered fragments, either terminal tails or surface loops. Although they often form instable and transient interactions, these fragments play essential roles in regulating macromolecular association or controlling the architecture of supramolecular complexes. The role of their conformational variability in complex formation is poorly understood and requires the development of specific approaches.
Methodology & Theoretical Orientation: We have studied the effect of protein segment conformational variability in protein-protein complex formation as well as peptide docking using theoretical docking approaches. Notably, we have developed a flexible docking method that accounts for the presence of flexible loops, together with analysis protocols that capture the entropic effects associated to structural variability in flexible docking results.
Findings: Whether the flexible segment is a loop or a peptide, we have found that a given mode of association can be stabilized by different conformations of the segment. Alternatively, different loop conformations can stabilize different modes of protein-protein association.
Conclusion & Significance: Tolerance of a binding site to conformational variability, as observed in protein-peptide docking but also in the association of proteins with flexible loops or segments, can play a role in adding a conformational entropy component to the energy of association, thus favoring the initial binding of the flexible fragment to its binding site. For proteins that associate using different binding geometries, either with different partners or along a functional pathway, loop flexibility can also be used to regulate the choice of the binding geometry.
References:
- Sacquin-Mora S, Prévost C (2015) Docking peptides on proteins: How to open a lock, in the dark, with a flexible key. Structure, 23:1373-1374.
- Laurin Y, Savarin P, Robert C, Takahashi M, Eyer J, Prévost C, Sacquin-Mora S (2015) Investigating the binding modes of the glioma targeting NFL-TBS.40-63 peptide on tubulin via docking and molecular dynamics simulations. Biochemistry 54:3660-3669.
- Bastard K, Thureau A, Lavery R and Prévost C (2003) Docking macromolecules with flexible segments. Journal of Computational Chemistry, 24:1910-1920.
- Bastard K, Prévost C and Zacharias M (2006) Accounting for loop flexibility during protein-protein docking. Proteins: Structure, Function and BioInformatics, 62:956-969
- Boyer B, Ezelin J, Poulain P, Saladin A, Zacharias M, Robert CH, Prévost C (2015). An integrative approach to the study of filamentous oligomeric assemblies, with application to RecA. PLoS ONE 10(3):e0116414.
- Track 3: 3D Structure Determination | Track 4: Computational Approaches | Track 5: Structural Molecular Biology
Location:
Session Introduction
Kurt Ballmer-Hofer
Paul Scherrer Institut, Switzerland
Title: Structural analysis of Vascular Endothelial Growth Factor receptors reveals drug-targetable allosteric sites regulating angiogenesis

Biography:
Kurt Ballmer-Hofer focused his research at PSI on the structural and functional analysis of receptor tyrosine kinases, in particular on Vascular Endothelial Growth Factor Receptors, VEGFRs. In collaboration with partner labs his team solved the structures of VEGF ligands, the ligand binding domain of VEGFR-2, and -3, and of the full-length extracellular domain of VEGFR-1 in complex with VEGF. The data of these studies led to the discovery of allosteric receptor regulatory sites in subdomains 4, 5 and 7. Antibodies and DARPins specifically binding to these domains showed strong inhibition of receptor activation and downstream signaling both in vitro and in vivo in angiogenesis model systems.
Abstract:
Vascular Endothelial Growth Factors (VEGFs) regulate blood and lymph vessel development upon activation of three receptor tyrosine kinases (RTKs), VEGFR-1, -2, and -3. Partial structures of VEGFR/VEGF complexes based on single particle electron microscopy, small angle X-ray scattering, and X-ray crystallography revealed the location of VEGF binding and the spatial arrangement of individual receptor subdomains. Here we describe the structure of the full-length VEGFR-1 extracellular domain (ECD) in complex with VEGF-A at 4 Å resolution. We combined X-ray crystallography, single particle electron microscopy, and molecular modeling for structure determination and validation. The structure reveals the molecular details of ligand-induced receptor dimerization, in particular of homotypic receptor interactions in Ig-domains 4, 5, and 7. Functional analyses of ligand binding and receptor activation confirm the relevance of these homotypic contacts for receptor activation and identify them as allosteric regulatory sites of VEGFR-1.
Based on our structural data we also investigated the function of Ig-domains 4, 5 and 7 in VEGFR-2, the primary receptor driving angiogenesis and vasculogenesis in response to VEGF administration. The basic domain structure of VEGFR-2 is very similar to VEGFR-1, the ECD of both receptors consists of 7 Ig-domains, D1-D7. Mutagenesis studies based on the VEGFR-1structure confirmed that Ig-domains 4 and 7 fulfill an essential regulatory function in receptor activation and may thus represent putative targets for pharmacological intervention. We isolated highly specific antibodies and DARPins (Designed Ankyrin Repeat Proteins) specific for domains 4 or 7. A subset of these reagents efficiently blocked receptor activation and inhibited VEGF-dependent signaling in vitro in endothelial cell cultures. Most importantly, a domain 4-specific DARPin efficiently blocked vessel development also in vivo in a mouse angiogenesis model. In this model endothelial cell spheroids were implanted in matrigel into mice, and cell growth and vessel formation were monitored in the absence and presence of inhibitor. Our study thus revealed a novel approach for therapeutic targeting of aberrant blood vessel development.
References:
- Markovic-Mueller, S., Stuttfeld, E., Asthana, M., Weinert, T., Bliven, S., Goldie, K.N., Kisko, K., Capitani, G., and Ballmer-Hofer, K. (2017). Structure of the Full-length VEGFR-1 Extracellular Domain in Complex with VEGF-A. Structure 25, 341-352.
- Leppänen, V.M., Tvorogov, D., Kisko, K., Prota, A.E., Jeltsch, M., Anisimov, A., Markovic-Mueller, S., Stuttfeld, E., Goldie, K.N., Ballmer-Hofer, K., and Alitalo, K. (2013). Structural and mechanistic insights into VEGF receptor 3 ligand binding and activation. Proc. Natl. Acad. Sci. USA 110, 12960-12965.
- Brozzo, M.S., Bjelic, S., Kisko, K., Schleier, T., Leppänen, V.M., Alitalo, K., Winkler, F.K., and Ballmer-Hofer, K. (2012). Thermodynamic and structural description of allosterically regulated VEGF receptor 2 dimerization. Blood 119, 1781-1788.
- Hyde, C.A., Giese, A., Stuttfeld, E., Abram, S.J., Villemagne, D., Schleier, T., Binz, H.K., and Ballmer-Hofer, K. (2012). Targeting the extracellular domains D4 and D7 of VEGFR-2 reveals allosteric receptor regulatory sites. Mol. Cell Biol. 32, 3802-3813.
- Leppänen, V.M., Prota, A.E., Jeltsch, M., Anisimov, A., Kalkkinen, N., Strandin, T., Lankinen, H., Goldman, A., Ballmer-Hofer, K., and Alitalo, K. (2010). Structural determinants of growth factor binding and specificity by VEGF receptor 2. Proc. Natl. Acad. Sci. USA 107, 2425-2430.
Biography:
Jianyong Li is a biochemistry professor at Virginia Tech. He has extensive experience in protein-related studies, including protein expression and purification, protein functional determination and protein structure and function relationships. Particularly worth to mention is the functional establishment of some unique yellow genes in insects and structural and function relationship of enzymes involved in kynurenate synthesis in mammals.
Abstract:
Bacterial effectors are proteins secreted by pathogenic bacteria into host cells through a type 3 or 4 secretion system. These bacterial effectors may help the pathogens to invade host cells and/or suppress its immune system; thereby promoting their infection, survival and reproduction. Effector proteins are primarily responsible for the pathogenicity of a given bacterial pathogen; therefore, learning the specific mechanism but which their effectors enter into host cells may provide insights for disease prevention. Bacterial Type 3 secretion system (T3SS) has been extensively studied. It is a needle-like structure made by a number of structural proteins, which is responsible for transfer of protein effector to host cells. The needle tip is ~ 3 nm, which is smaller than the required dimension for most bacterial effectors. Despite some better understanding of the T3SS structure, how their bacterial effectors gain entry to host cells remains speculative. Using a T3SS-dependant effector as a model from Xanthomonas oryzae (a bacterium causing serious disease to some essential plants), we determined the structures of an effector protein in complex with a chaperone-like protein. In the genome of Xanthomonas oryzae, the coding sequence of effector protein is adjacent to that coding the chaperone-like protein. Our structural analysis indicates that the effector-chaperon complex crystallized as tetramers (1.64 A resolution). The monomer of the protein effector contains a T4 polynucleotide kinase domain, while the monomer of the chaperon includes a novel kinase binding domain. Our data suggest that the chaperone protein interacts with the protein effector in a manner that helps to stabilize the protein effector and prevents the virulence effect of protein effort from harming the bacteria before being transferred to host cells. Currently, efforts are being made to understand the precise roles the chaperon protein plays during the transfer of protein effector to host cells.
References:
- Han et al. (2017) Crystal structure of acetylcholinesterase subunits of the malaria vector Anopheles gambiae. Insect Science (https://www.ncbi.nlm.nih.gov/pubmed/28247978).
- Carlier PR, Bloomquist JR, Totrov M and Li J (2017) Discovery of species-selective and resistance-breaking anticholinesterase insecticide for the malaria mosquito. Current Medical Chemistry (https://www.ncbi.nlm.nih.gov/pubmed/28176636)
- Yang et al. (2016) Kynurenine aminotransferase 3/glutamine transaminase L/cysteine conjugate beta-lyase 2 is a major glutamine transaminase in the mouse kidney. Biochemistry and Biophysics Reports 8: 231-241.
- Han Q et al. (2015) crystal structure of the complex between AvrRxo1-ORF1, a type III effector and its cognate chaperon, AvrRxo1-ORF2. Structure 23: 1900-1909 (http://www.sciencedirect.com/science/article/pii/S0969212615003251).
- Han Q, Robinson H, Ding H, Christensen BM and Li J (2012) Evolution of insect arylalkylamine N-acetyltransferases: structural evidence from the yellow fever mosquito, Aedes aegypti. Proceedings of the National Academy of Sciences 109: 11669-11674.

Biography:
Kyoko Shinzawa-Itoh, associate Professor of University of Hyogo, grew up in Hiroshima Japan. She received her MS degree from Hiroshima University of Graduate School of Integrated Arts and Sciences and her Ph D. in Pharmaceutical Sciences from Hiroshima University of Graduate School of Biomedical & Health Sciences. She worked as assistant professor at the Department of Life Science, Himeji Institute of Technology 1988-2004 and at the Hyogo University of Graduate School of Life Science 2004-2013. She is associate Professor of Picobiology Institute, Graduate School of Life Science University of Hyogo from 2013. She has studied about mitochondrial respiratory complexes.
Abstract:
Mitochondrial cytochrome c oxidase (CcO) transfers electrons from cytochrome c (Cyt.c) to O2 to generate H2O, a process coupled to proton pumping. To elucidate the mechanism of electron transfer, a crystal structure of the complex of CcO and Cyt.c would be invaluable for mechanistic studies. Two-dimensional (2D) crystals of the mammalian Cyt.c–CcO complex were prepared at higher pH (7.4–9.0) with both proteins in the oxidized state (Osuda et al, 2016), but these 2D crystals could not provide us with a structure of sufficient resolution. We optimized 3D crystallization conditions for ferri-Cyt.c and oxidized CcO at high pH and solved the X-ray structure of the complex at 2.0 Å resolution. The specific interaction between Cyt.c and CcO is stabilized by only six electrostatic interactions between side chains within a small contact surface. From a theoretical calculation based on the complex structure, we identified an electron transfer pathway from the heme c of Cyt.c to CuA of CcO via Lys-13 of Cyt.c. Between the two proteins are three water layers, one of which lies between the other two layers without significant direct interaction with either protein. The inter-molecular span between Cyt.c and CcO is longer than those of other complexes by more than 3.0 Å, and the contact surface area of Cyt.c and CcO is smaller than one-third the size of those of other complexes. Cyt.c undergoes large structural fluctuations, using the interacting regions with CcO as a fulcrum. These features of the protein–protein interaction at the docking interface represent the first known example of a new class of inter-protein interaction, which we term “soft and specific”. This interaction is predicted to contribute to the rapid association/dissociation of the Cyt.c–CcO complex, which facilitates the sequential supply of four electrons for the O2 reduction reaction.
References:
- Osuda Y, Shinzawa-Itoh K, Tani K, Maeda S, Yoshikawa S, Tsukihara T, Gerle C (2016) Two-dimensional crystallization of monomeric bovine cytochrome c oxidase with bound cytochrome c in reconstituted lipid membranes. Microscopy. 65(3):263-267.
- Shimada S, Shinzawa-Itoh K, Baba J, Aoe S, Shimada A, Yamashita E, Kang J, Tateno M, Yoshikawa S, Tsukihara T (2017) Complex structure of cytochrome c-cytochrome c oxidase reveals a novel protein-protein interaction mode. EMBO J. 36(3):291-300.
- Shinzawa-Itoh K, Shimomura H, Yanagisawa S, Shimada S, Takahashi R, Oosaki M, Ogura T, TsukiharaT (2016) Purification of active respiratory supercomplex from bovine heart mitochondria enables functional studies. J. Biol. Chem. 291(8):4178-84.
- Hirata K, Shinzawa-Itoh K, Yano N, Takemura S, Kato K, Hatanaka M, Muramoto K, Kawahara T, Tsukihara T, Yamashita E, Tono K, Ueno G, Hikima T, Murakami H, Inubushi Y, Yabashi M, Ishikawa T, Yamamoto M, Ogura T, Sugimoto H, Shen JR, Yoshikawa S, Ago H (2014) Determination of damage-free crystal structure of an X-ray-sensitive protein using an XFEL. Nat Methods. 11(7):734-736.
Jeffrey L Urbauer
The University of Georgia, USA
Title: Oxidative stress, methionine oxidation, and calmodulin structure and function

Biography:
Jeffrey Urbauer earned bachelor’s and doctoral degrees in chemistry from the University of Nebraska-Lincoln. He pursued postdoctoral studies at the University of Wisconsin-Madison as an NIH postdoctoral fellow and at the University of Illinois Urbana-Champaign. He held faculty appointments at the State University of New York at Buffalo, the University of Pennsylvania, and the University of Kansas before joining the faculty in the Department of Chemistry and the Department of Biochemistry and Molecular Biology at the University of Georgia. At the University of Kansas the Mortar Board National College Senior Honor Society awarded him with the Outstanding Educator Award. His research interests include structural biology, protein biophysics and NMR spectroscopy.
Abstract:
Statement of the Problem: Oxidation of methionine residues in proteins to methionine sulfoxide is a prevalent, reversible post-translational modification. Changes in protein structure and function accompany oxidation due to polarity and steric differences between methionine and the sulfoxide. We are investigating the consequences of methionine oxidation in the regulatory protein calmodulin (CaM), a key calcium signal transducer with nine methionine residues, in hydrophobic pockets of its opposing globular domains, which interact with target proteins. CaM with oxidized methionine residues accumulates under conditions of oxidative stress, and because of its central role in biology, it is important to understand the functional effects of these alterations and their physical origins. Methodology: Methionine residues in CaM are easily oxidized in vitro with hydrogen peroxide. To study the effects of oxidation of specific methionine residues, leucine was substituted for methionine at remaining sites. A combination of functional assays, single molecule studies, and NMR spectroscopy were used to assess functional and structural consequences of methionine oxidation. Findings: For the best studied case, activation of the plasma membrane Ca-ATPase (PMCA) by CaM, impaired CaM function is due to oxidation of a single C-terminal methionine. Single molecule experiments indicate non-productive binding of oxidized CaM to the PMCA. High resolution NMR studies demonstrate significant structural perturbation in the C-terminal globular domain of oxidized CaM and an inability to anchor the PMCA to this domain. Conclusion & Significance: The functional effects of methionine oxidation in CaM are highly target dependent, as is the degree to which selective oxidation of particular methionine residues in CaM affects function. The results of CaM activation of the PMCA also indicate that both high-affinity productive and non-productive complexes of oxidized CaM with targets are possible. These facts indicate that a comprehensive understanding of the metabolic consequences of CaM oxidation will be challenging.
References:
- Bartlett RK, Bieber Urbauer RJ, Anbandam A, Smallwood HS, Urbauer JL, Squier TC (2003) Oxidation of Met144 and Met145 in calmodulin blocks calmodulin dependent activation of the plasma membrane ca-ATPase. Biochemistry 42:3231-3238.
- Osborn KD, Bartlett RK, Mandal A, Zaidi A, Bieber Urbauer RJ, Urbauer JL, Galeva N, Williams TD, Johnson CK (2004) Single-molecule dynamics reveal an altered conformation for the autoinhibitory domain of plasma membrane Ca-ATPase bound to oxidatively modified calmodulin. Biochemistry 43:12937-12944.
- Anbanandam A, Bieber Urbauer RJ, Bartlett RK, Smallwood HS, Squier TC, Urbauer, JL (2005) Mediating molecular recognition by methionine oxidation: Conformational switching by oxidation of methionine in the carboxyl-terminal domain of calmodulin. Biochemistry 44:9486-9496.
- Slaughter BD, Bieber Urbauer RJ, Urbauer JL, Johnson CK (2007) Mechanism of calmodulin recognition of the binding domain of isoform 1b of the plasma membrane Ca-ATPase: Kinetic pathway and effects of methionine oxidation. Biochemistry 46: 4045-4054.
- Lubker C, Bieber Urbauer RJ, Moskovitz J, Dove S, Weisemann J, Fedorova M, Urbauer JL, Seifert R (2015) Membranous adenylyl cyclase 1 activation is regulated by oxidation of N- and C-terminal methionine residues in calmodulin. Biochemical Pharmacology 93:196-209.
Miki Senda
High Energy Accelerator Research Organization (KEK), Japan
Title: Molecular mechanism of SHP2 activation by CagA from Helicobacter pylori

Biography:
Miki Senda has completed her PhD at 2008 from Nagaoka University of Technology. She is an assistant professor of Structural Biology Research Center in High Energy Accelerator Research Organization (KEK). She has several collaborations, in which she has worked as an expert of protein crystallization and crystal quality improvement. She received Oxford Cryosystems Low Temperature Prize at the 63rd Annual meeting of the American Crystallographic Association (ACA) in 2013.
Abstract:
Helicobacter pylori, which is known as a major risk factor of stomach cancer, delivers an effector protein CagA into gastric epithelial cells. CagA then promiscuously interact with host proteins, SHP2 and PAR1b, to deregulate these proteins, potentiating oncogenic signaling. CagA comprises an N-terminal structured region and a C-terminal intrinsically disordered region which interacts with the host proteins. We already determined the crystal structure of the N-terminal region of CagA (1–3). The crystal structure revealed that the basic amino-acid cluster in the N-terminal region is utilized to localized CagA at the inner face of the plasma membrane. After localization, short segments including Glu-Pro-Ile-Tyr-Ala (EPIYA) motif in the C-terminal disordered region are phosphorylated by Src and interact with SHP2 to deregulate its phosphatase activity. In this study, we have analyzed the structure-function relationship of the EPIYA-segments of CagA. Based on the sequence flanking each of the EPIYA motifs, four types of EPIYA segments, A, B, C, and D, have been identified (4). It is already known that combinations of the EPIYA segments are geographically different (Western and East Asian CagA) and affect CagA’s oncogenic activity. While Western CagA with EPIYA-A, B, and C segments has weak oncogenic activity, Western CagA with EPIYA-A, B and multiple EPIYA-C segments shows increased oncogenic activity (5). East Asian CagA, which has much higher oncogenic activity than Western ones, typically possesses EPIYA-A, B, and D in the C-terminal region. Our biochemical data revealed that oncogenic activity of CagA is correlated with binding affinity for SH2 domain of SHP2 (SH2_SHP2). We have analyzed the interaction between SH2_SHP2 and the EPIYA-C/D segment using biochemical, crystallographic, and physicochemical methods and revealed two types of activation mechanisms of SHP2. In our presentation, we will report that East Asian and Western CagA utilize two distinct activation mechanisms of SHP2.
References:
- Hayashi T et al., (2012) Tertiary structure-function analysis reveals the pathogenic signaling potentiation mechanism of Helicobacter pylori oncogenic effector CagA. Cell host & microbe 12:20-33.
- Senda M et al., (2016) Use of multiple cryoprotectants to improve diffraction quality from protein crystals. Crystal growth & design 16: 1565-1571.
- Senda M (2016) A comprehensive strategy to obtain high quality crystals. Structural Biology 2016
- Hatakeyama M (2004) Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat. Rev. Cancer 4: 688-694.
- Nagase L et al., (2015) Dramatic increase in SHP2 binding activity of Helicobacter pylori Western CagA by EPIYA-C duplication: its implications in gastric carcinogenesis. Sci. Rep. 5: 15749.
Alfredo De Biasio
Elettra-Sincrotrone Trieste, Italy
Title: Integrative approaches to study the structure and motions of DNA sliding clamps
Time :

Biography:
Alfredo De Biasio’s work focuses on the structure and function of DNA sliding clamps and their complexes operating in DNA replication and repair. He is particularly interested in understanding the mechanisms of sliding of the eukaryotic clamp PCNA, and how these mechanisms are modulated by modifications of the PCNA sliding surface, and the implications in DNA damage avoidance. These problems are tackled by an integrative approach that combines X-ray crystallography, NMR and MD simulations.
Abstract:
Sliding clamps encircle DNA and tether polymerases and other proteins to the genomic template, and are essential factors in DNA replication. Because of the transient interaction that the clamps establish with DNA, the clamp-DNA interface eluded a thorough structural characterization, so that the molecular mechanism for clamp sliding on DNA remained obscure. Here, I will show how the combined use of high-resolution techniques (X-ray crystallography and NMR) and molecular dynamics (MD) simulations allowed to visualize the interactions between the Proliferating Cell Nuclear Antigen (PCNA) – the eukaryotic sliding clamp – and DNA, and to decipher the mechanics of sliding. In addition, recent findings show that the DNA sliding surface of PCNA can be modified to regulate the resistance to DNA damage. From a structural viewpoint, I will reflect on these findings which open a new perspective on PCNA function and offer opportunities to develop tools to manipulate the DNA damage response in cancer treatment.
References:
1. De March, M. et al. Structural basis of human PCNA sliding on DNA. Nat. Commun. 8, 13935 (2017).
2. De Biasio, A. et al. Structure of p15PAF‒PCNA complex and implications for clamp sliding during DNA replication and repair. Nat. Commun. 6, 6439 (2015).

Biography:
Liliane Mouawad was always interested in understanding the mechanism of action of proteins or protein assemblies. This understanding may be based on either molecular simulations or on experiments like NMR. But her expertise is primarily in molecular dynamics simulations and more precisely in normal mode analysis (NMA). She has developed several methods going from the calculation of normal modes of very large systems or of images, to the calculation of the pathway between two protein conformations, to the prediction of the compactness of a calcium-binding protein. Recently she was also involved in docking and virtual screening themes, where she has acquired enough expertise to develop a new consensus methodology to overcome some issues observed in these approaches.
Abstract:
A microtubule is a dynamic system formed of ab-tubulins. The presence of nonhydrolyzable guanosine-5’-triphosphate (GTP)/guanosine diphosphate (GDP) on the b-tubulins provokes microtubule polymerization/depolymerization. Despite the large number of experimental studies of this dynamical process, its mechanism is still unclear. To provide insights into this mechanism, we studied the first depolymerization steps of GDP/GTP-bound microtubules by normal-mode analysis with the all-atom model. We also constructed a depolymerizing microtubule and compared it to cryo-electron microscopy tomograms (cyro-ET). The results show that during depolymerization, the protofilaments not only curve but twist to weaken their lateral interactions. These interactions are stabilized by GTP, but not evenly. Not all of the interface residues are of equal importance: five of them, belonging to the H2-S3 loop, play a special role; acting as a lock whose key is the g-phosphate of GTP. Sequence alignments of several tubulins confirm the importance of these residues.
References:
- Chaput L, Martinez‑Sanz J, Saettel N, Mouawad L (2016) Benchmark of four popular virtual screening programs: construction of the active/decoy dataset remains a major determinant of measured performance. J Cheminform 8:56-72.
- Chaput L, Martinez‑Sanz J, Quiniou E, Rigolet P, Saettel N, Mouawad L (2016) vSDC: a method to improve early recognition in virtual screening when limited experimental resources are available. J Cheminform 8:1-18.
- Quiniou E, Guichard P, Perahia D, Marco S, Mouawad L (2013) An atomistic view of microtubule stabilization by GTP Structure 21: 833–843.
- Martinez-Sanz J, Kateb F, Assairi L, Blouquit Y, Bodenhausen G, Abergel D, Mouawad L, Craescu CT (2010) Structure, dynamics and thermodynamics of the human centrin 2/hSfi1 complex. J. Mol. Biol. 395: 191–204.
- Mouawad L, Isvoran A, Quiniou E, Craescu CT (2009) What determines the degree of compactness of a calcium-binding protein? FEBS Journal 276:1082–1093.
Hyun-Soo Cho
Yonsei University, Korea
Title: Structure and function of a Chloride Pump Rhodopsin from marine bacteria

Biography:
Hyun-Soo Cho’s research aims to understand the structural and functional role of various proteins involved in cancer and immune diseases. He is specialized in X-ray Crystallography to solve protein structures with other biophysical and biochemical techniques including Cryo_EM recently. His ongoing research projects include various enzymes and receptors especially G-Protein Coupled Receptor (GPCR) related with cancer and immune system.
Abstract:
Recently, light-driven sodium pump rhodopsin (NaR/KR2/NDQ rhodopsin) and chloride pump rhodopsin (ClR/NTQ rhodopsin) from marine flavobacteria were identified by metagenomics study. One of them, light-driven sodium pump rhodopsin (NaR) structure was determined. The other one we have solved the first crystal structure of a unique class light-driven chloride pump (ClR) from Nonlabens marinus S1-08, at resolutions of 1.57 Å. Like structured Halorhodopsin (HR), ClR can transfer chloride ion from extracellular to cytosol. Although both ClR and HR are same light-driven chloride pump rhodopsin, we found some evidences that ClR and HR are different in structure and mechanism. The structures reveal two chloride-binding sites, one around the protonated Schiff base and the other on a cytoplasmic loop. We identify a “3 omega motif” formed by three non-consecutive aromatic amino acids that is correlated with the B-C loop orientation. Detailed CIR structural analyses with functional studies in E. coli reveal the chloride ion transduction pathway. Our results help understand the molecular mechanism and physiological role of ClR and provide a structural basis for optogenetic applications.
References:
- Kim K, Kwon SK, Jun SH, Cha JS, Kim H, Lee W, Kim J*, Cho HS*, Crystal structure and functional characterization of a light-driven chloride pump having an NTQ motif. Nature Communications, 7, 12677 (2016)
- Lim Y, Yoo J, Kim MS, Hur M, Lee EH, Hur HS, Lee JC, Lee SN, Park TW, Lee K, Chang KH, Kim K, Kang YJ, Hong KW, Kim SH, Kim YG, Yoon Y, Nam DH, Yang H, Kim DG, Cho HS*, Won J*, GC1118, an Anti-EGFR Antibody with a Distinct Binding Epitope and Superior Inhibitory Activity against High-Affinity EGFR Ligands. Molecular Cancer Therapeutics, 15, 251-263 (2016)
- Lee J, Choi HJ, Yun M, Kang YJ, Jung JE, Ryu Y, Kim TY, Cha YJ, Cho HS*, Min JJ*, Chung CW*, Kim HS*, Enzymatic Prenylation and Oxime Ligation for the Synthesis of Stable and Homogeneous Protein-Drug Conjugates for Targeted Therapy. ANGEWANDTE CHEMIE-INTERNATIONAL EDITION, 54, 12020-12024 (2015)
- Jeong SA, Kim K, Lee JH, Cha JS, Khadka P, Cho HS*, Chung IK*, Akt-mediated phosphorylation increases the binding affinity of hTERT for importin α to promote nuclear translocation. Journal of Cell Science, 128, 2287-2301 (2015)
- Cho YS, Yoo J, Park S, Cho HS*, The structures of the kinase domain and UBA domain of MPK38 suggest the activation mechanism for kinase activity. Acta Crystallography D, 70, 514-521 (2014)
Stanislav Engel
Ben-Gurion University, Israel
Title: Construction of Structural Mimetics of the Thyrotropin Receptor Intracellular Domain

Biography:
Stanislav Engel, Ph.D., now is an Assistant Professor in the Department of Clinical Biochemistry and Pharmacology, Faculty of Health Sciences, The National Institute for Biotechnology, Ben-Gurion University in the Negev, Beer-Sheva, Israel. He got his B Sc in biochemistry, M Sc and Ph.D. in biochemistry and biotechnology engineering at the Ben-Gurion University in the Negev. Currently, Dr. Stanislav Engel’ researches focus on the understanding the structural basis of “protein misfolding” diseases, such as ALS, and structure-based drug discovery.
Abstract:
Dissecting G protein-coupled receptors (GPCR) signaling in terms of the pathways being activated will boost our understanding of the molecular fundamentals of hormone action. The structural determinants governing the selectivity of GPCR/G protein coupling, however, remain obscure. The selectivity of GPCR/G protein recognition appears to be determined by both specific inter-residue interactions and features related to the overall 3D conformation of the ICD. It appears, therefore, that to elucidate the fundamentals of the selectivity of GPCR/G protein recognition, a comprehensive analysis of the structure-activity relationships of multiple GPCR complexes with different G protein isoforms is required. However, enormous technical difficulties associated with the isolation of functional receptors in quantities required for direct structural studies effectively impede progress in the field. Methodology: We constructed the functional mimetics of the intracellular domain (ICD) of a model GPCR - thyrotropin receptor (TSHR), based on a unique scaffold, 6-Helix, an artificial protein that was derived from the elements of the trimer-of-hairpins structure of HIV gp41 and represents a bundle of six a-helices. Findings: The 6-Helix scaffold, which endowed the substituted TSHR ICD elements with spatial constraints analogous to those, found in native receptors, enabled the reconstitution of a microdomain comprising the intracellular loops ICL-2 and ICL-3, which is capable of binding and activating Ga-(s). Conclusion & Significance: By using a soluble scaffold, which furnishes peptides derived from the GPCR ICD with spatial constraints similar to those, found in native receptors, the reconstitution of a native-like G protein-recognition epitope can be facilitated. The 6-Helix-based mimetics could be used as a platform to study the molecular basis of GPCR/G protein recognition. Such knowledge could lead to the development of novel therapeutic strategies for GPCR-related disorders by targeting the GPCR/G protein interfaces and help counteract cellular dysfunctions via focused tuning of GPCR signaling.
References:
- Y. Y. Kuttner and S. Engel, Protein hot spots - the islands of stability, Journal of Molecular Biology, 415(2):419-28 (2012)
- Y. Y. Kuttner, T. Nagar and S. Engel, Surface dynamics in allosteric regulation of protein-protein interactions: Modulation of calmodulin functions by Ca2+, PLOS Computational Biology, 9(4): e1003028 (2013)
- B. Katzman, M. Vyazmensky, O. Press, M. Volokita and S. Engel, Tethered ribozyme ligation enables detection of molecular proximity in homogeneous solutions, Biotechnology Journal, 10 (3):379–385 (2015)
- A. Kuzmina, K. Vaknin, G. Gdalevsky, M. Vyazmensky, R.S. Marks, R. Taube and S. Engel, Functional Mimetics of the HIV-1 CCR5 Co-Receptor Displayed on the Surface of Magnetic Liposomes, PLOS One, 10(12): e01440432015 (2015)
- R. Osman, M. Mezei and S. Engel, The role of protein “Stability patches” in molecular recognition: A case study of the human growth hormone-receptor complex, Journal of Computational Chemistry, 37 (10):913-919 (2016)
Mi Sun Jin
GIST, Republic of Korea
Title: Structural Basis of Sodium/Citrate Symporter as a Secondary Transporter

Biography:
Mi Sun Jin - Assistant Professor, 2014-Present, School of Life Sciences, GIST;
Education - B.S. 2002, Sogang University; M.S. 2004, KAIST; Ph.D. 2008, KAIST (advisor: Jie-Oh Lee)
Professional Experience - Postdoctoral Fellow, 2008-2009, KAIST (advisor: Jie-Oh Lee),
Postdoctoral Fellow, 2009-2013, Purdue University (advisor: Jue Chen),
Research Specialist, 2013-2014, Purdue University
Abstract:
The sodium-dependent citrate transporter of Klebsiella pneumoniae (KpCitS) belongs to the 2-hydroxycarboxylate transporter (2-HCT) family and allows the cell to use citrate as sole carbon and energy source in anaerobic conditions. We present crystal structures of KpCitS in its citrate-bound outward-facing as well as citrate-free inward-facing state. The structure of the asymmetric KpCitS homodimer containing both outward- and inward-open protomers was also determined. The structures reveal that the KpCitS dimerization domain remains stationary throughout the transport cycle due to an extensive hydrogen bond network as well as hydrophobic interactions. In contrast, its transport domain undergoes a ~35° rigid-body rotation and a ~17 Å translocation perpendicular to the membrane to expose the substrate-binding site alternately to either side of the membrane. Homology models of two other 2-HCT proteins based on the KpCitS structure offer structural insights into their differences in substrate specificity at a molecular level. On the basis of our results and previous biochemical data, we propose that the activity of the 2-HCT family of transporters involves an elevator-like movement in which the transport domain itself traverses the lipid bilayer, carrying the substrate into the cell in a sodium-dependent manner.
References:
- Kim JH, Song DH, Youn SJ, Kim JW, Cho G, Kim SC, Lee H, Jin MS, Lee JO (2016) Crystal structures of mono- and bi-specific diabodies and reduction of their structural flexibility by introduction of disulfide bridges at the Fv interface. Scientific reports, 6, 34515.
- Mi Sun Jin, Michael L. Oldham, Qiuju Zhang, Jue Chen (2012) Crystal structure of the multidrug transporter P-glycoprotein from C. elegans. Nature, 490, 566-9.
- Jin Young Kang, Xuehua Nan, Mi Sun Jin, Suk-Jun Youn, Young Hee Ryu, Shinji Ma, Seung Hyun Han, Sang-Gi Paik, Hayyoung Lee, Jie-Oh Lee (2009) Recognition of lipopeptide patterns by TLR2-TLR6 heterodimer. Immunity, 31, 873-84.
- Mi Sun Jin, Jie-Oh Lee (2008) Structures of the Toll-like Receptor Family and Its Ligand Complexes. Immunity, 29, 182-91.
- Mi Sun Jin, Sung Eun Kim, Jin Young Heo, Mi Eun Lee, Ho Min Kim, Sang-Gi Paik, Hayyoung Lee, Jie-Oh Lee (2007) Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a triacylated lipopeptide. Cell, 130, 1071-82.
Pedro J. de Pablo
Universidad Autonoma de Madrid, Spain.
Title: Atomic Force Microscopy of virus capsids uncover the interplay between mechanics, structure and function

Biography:
Pedro J. de Pablo has his expertise in the application of Atomic Force Microscopy to the investigation of individual viruses. He studied Condensed Matter Physics at the Universidad Autonoma de Madrid and worked with a variety of molecular wires, including carbon nanotubes and DNA during his PhD. Then he moved for a postoc in the Vrije Universiteit to apply AFM to the study of molecular motors (kinesin) and viruses. He stablished his own group back in Madrid, where during the last 10 years has been applying AFM to unveil the structure-function interplay of viruses by measuring thir physical properties, such as mechanics and electrostatics.
Abstract:
The basic architecture of a virus consists of the capsid, a shell made up of repeating protein subunits, which packs, shuttles and delivers their genome at the right place and moment. Viral particles are endorsed with specific physicochemical properties, which confer to their structures certain meta-stability whose modulation permits fulfilling each task of the viral cycle. These natural designed capabilities have impelled using viral capsids as protein containers of artificial cargoes (drugs, polymers, enzymes, minerals) with applications in biomedical and materials sciences. Both natural and artificial protein cages (1) have to protect their cargo against a variety of physicochemical aggressive environments, including molecular impacts of highly crowded media, thermal and chemical stresses, and osmotic shocks. Viral cages stability under these ambiences depend not only on the ultimate structure of the external capsid, which rely on the interactions between protein subunits, but also on the nature of the cargo. During the last decade our lab has focused on the study of protein cages with Atomic Force Microscopy (AFM) (figure 1). We are interested in stablishing links of their mechanical properties with their structure and function. In particular, mechanics provide information about the cargo storage strategies of both natural and virus-derived protein cages (2,3). Mechanical fatigue has revealed as a nanosurgery tool to unveil the strength of the capisd subunit bonds (4). We also interrogated the electrostatics of individual protein shells (5). Our AFM-fluorescence combination provided information about DNA diffusing out cracked-open protein cages in real time (6).
References:
- Llauró et al. Nanoscale, 2016, 8, 9328.
- Hernando-Pérez et al. Small, 2012, 8, 2336.
- Ortega-Esteban et al. ACS Nano, 2015, 9, 10826.
- Hernando-Pérez et al. Nature Communications, 2014, 5, 4520.
- Hernando-Pérez et al. Nanoscale, 2015, 7, 17289.

Biography:
The Kolaczkowski Lab uses a combination of computational biology, statistics, structural modeling and biochemistry to examine how molecular systems evolve. We are particularly interested in the evolution of innate immunity in animals and plants. Dr. Kolaczkowski’s research program focuses on developing cutting-edge tools for the characterization of molecular-evolutionary processes and applying these approaches to learn how long-term protein evolution works. Dr. Kolaczkowski’s recent work has advanced the fields of ancestral sequence resurrection (ASR) and protein function prediction while shedding new light on the evolution of innate antiviral immunity and RNA interference.
Abstract:
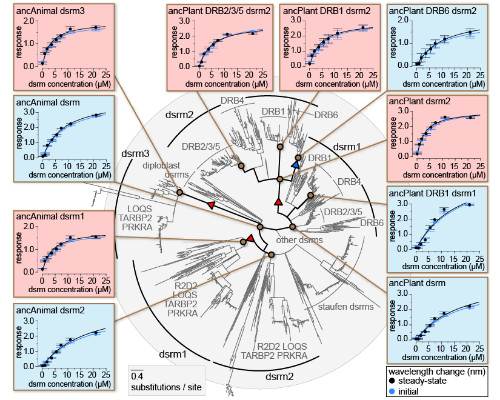
- Pugh C, Kolaczkowski O, Manny A, Korithoski B, Kolaczkowski B (2016) Resurrecting the ancestral structural dynamics of an antiviral immune receptor: Adaptive binding pocket reorganization repeatedly shifts RNA preference. BMC Evolutionary Biology 16(1):241.
- Dias R, Kolaczkowski B (2015) Different combinations of atomic interactions predict protein-small molecule and protein-DNA/RNA affinities with similar accuracy. Proteins 83(11):2100-2114.
- Korithoski B, Kolaczkowski O, Mukherjee K, Kola R, Earl C, Kolaczkowski B (2015) Evolution of a novel antiviral immune signaling interaction by partial-gene duplication. PLoS One 10(9):e0137276.
- Mukherjee K, Korithoski B, Kolaczkowski B (2014) Ancient origins of vertebrate-specific innate antiviral immunity. Molecular Biology and Evolution 31(1):140-153.
- Mukherjee K, Campos H, Kolaczkowski B (2013) Evolution of animal and plant Dicers: Early parallel duplications and recurrent adaptation of antiviral RNA binding in plants. Molecular Biology and Evolution 30(3):627-641.
John van Noort
Leiden University, The Netherlands
Title: The structure of chromatin; single-molecule experiments on model fibers and real genes

Biography:
Chromatin is the ubiquitous protein-DNA complex that forms the structural basis of DNA condensation in all eukaryotic organisms. Packaging and depackaging of chromatin, called chromatin remodeling, plays a central role in all cellular processes that involve chromosomes such as transcription, replication, recombination and repair. Detailed knowledge of the principles and mechanisms underlying this control of DNA condensation is thus vital for understanding many diseases, including neurological disorders and cancer. The physical mechanisms governing these processes however, are still largely unknown. I am interested in developing and using modern biophysical techniques to unravel the physics behind DNA condensation and its role in transcription regulation.
Abstract:
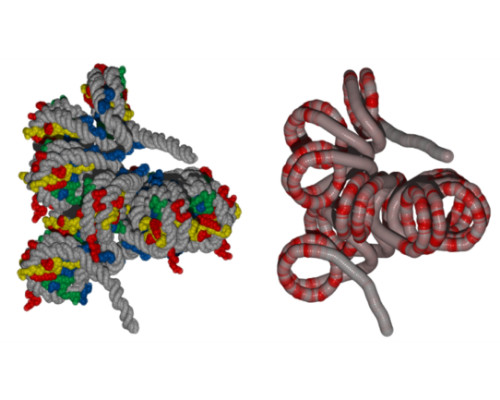
- Multiplexing genetic and nucleosome positioning codes: a computational approach (2016) B Eslami-Mossallam, RD Schram, M Tompitak, J van Noort, H Schiessel PloS one 11 (6), e0156905
- Quantitative analysis of single-molecule force spectroscopy on folded chromatin fibers (2016) H Meng, K Andresen, J Van Noort Nucleic acids research 43 (7), 3578-3590
- spFRET reveals changes in nucleosome breathing by neighboring nucleosomes (2015) R Buning, W Kropff, K Martens, J van Noort Journal of Physics: Condensed Matter 27 (6), 064103
- Histone H3 phosphorylation near the nucleosome dyad alters chromatin structure (2014) Justin A North, Marek Šimon, Michelle B Ferdinand, Matthew A Shoffner, Jonathan W Picking, Cecil J Howard, Alex M Mooney, John van Noort, Michael G Poirier, Jennifer J Ottesen Nucleic acids research 42 (8), 4922-4933
- Sequence-based prediction of single nucleosome positioning and genome-wide nucleosome occupancy (2012) T van der Heijden, JJFA van Vugt, C Logie, J van Noort Proceedings of the National Academy of Sciences 109 (38), E2514-E25225.
- Track 6: Frontiers in Structural Biology
Session Introduction
Petra Fromme
Arizona State University, USA
Title: Dynamics of Biomolecules “In Action†Studied with X-ray Free Electron Lasers

Biography:
Petra Fromme received her masters at the Free University in Berlin in Biochemistry (1985) and then received her doctorate in Chemistry at the Technical University in Berlin (1988) where she then became a professor in 1992. During this time she developed and pursued her fascination with understanding the function of membrane proteins by investigating and determining their atomic structures. In 2002, Dr. Fromme joined Arizona State University as a professor of Chemistry and Biochemsitry where she has worked with distinguished colleagues from around the world to pioneer a new technique for imaging proteins using extraordinarily powerful x-ray lasers that has the capability to make movies of these fascinating proteins in action.
Abstract:
Biomolecules are highly dynamic; however, most structures only provide a static picture of the molecule. Serial Femtosecond Crystallography (SFX) provides a novel concept for structure determination, where X-ray diffraction “snapshots” are collected from a fully hydrated stream of nanocrystals, using femtosecond pulses from high energy X-ray free-electron lasers (XFELs), where diffraction is observed before destruction takes effect [1]. The first proof of concept of serial femtosecond crystallography was achieved using Photosystem I, a larger membrane protein complex involved in Photosynthesis as a model system [2]. The structure of non-damaged biomolecules can now be determined, unraveling their function at the atomic scale [3],[4] that include important human membrane-bound receptors [6], [8] [9]. SFX opens a new avenue for determination of protein dynamics, with the goal of molecular movies of biomolecules “in action”. First experiments on the proof of principle for time resolved serial femtosecond nanocrystallography have been performed on proteins in Photosynthesis, where first snapshots of steps in water splitting reaction have been observed [10]. A new concept based on continuous X-ray diffraction extends resolution beyond Bragg diffraction and allows for direct phasing of X-ray diffraction data [11]. TR-SFX studies extend to atomic resolution where the first steps in photosensing were recently revealed at a time scale of femtoseconds using the photoactive yellow protein [12,13]. This pioneering work paves the way for the determination of molecular movies of the dynamics of membrane proteins "at work" in the future.
The talk will close with a progress report on the development of compact femto and attosecond X-ray Sources at DESY (AXSIS) and at ASU (CXLS and CXFEL) , which will provide unique new opportunities to study the ultrafast dynamics of reactions in photosynthesis with a combination of X-ray diffraction, X-ray spectroscopy and ultrafast optical spectroscopy [14].
References:
- Barty A, Caleman C, Aquila A, Timneanu N, Lomb L, et al. (2012) Self-terminating diffraction gates femtosecond X-ray nanocrystallography measurements. Nature Photonics 6: 35-40.
- Chapman HN, Fromme P, Barty A, White TA, Kirian RA, et al. (2011) Femtosecond X-ray protein nanocrystallography. Nature 470: 73-U81.
- Boutet S, Lomb L, Williams GJ, Barends TRM, Aquila A, et al. (2012) High-Resolution Protein Structure Determination by Serial Femtosecond Crystallography. Science 337: 362-364.
- Redecke L, Nass K, DePonte DP, White TA, Rehders D, et al. (2013) Natively Inhibited Trypanosoma brucei Cathepsin B Structure Determined by Using an X-ray Laser. Science 339: 227-230.
- Kang YY, Zhou XE, Gao X, He YZ, Liu W, et al. (2015) Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 523: 561-+.
Maria Bykhovskaia
Wayne State University, USA
Title: Protein Machinery Regulating the Synaptic Vesicle Fusion

Biography:
Maria Bykhovskaia is an expert in synaptic transmission. Her lab combines molecular modeling and computations with electrophysiology, microscopy, and molecular biology approaches. She hold a Professor’s position in the WSU. Her Ph.D. training was in protein molecular modeling, and subsequently she used a postdoc in computational neuroscience to initiate a career devoted to the study of presynaptic mechanisms and plasticity. As a PI, she has developed in her lab expertise in electrophysiology, live confocal imaging, and electron microscopy. The lab combines these experimental approaches with mathematical modeling to understand the fundamental mechanisms of release of neuronal transmitters.
Abstract:
Neuronal transmitters are released via the fusion of synaptic vesicles with the plasma membrane. Vesicles dock to the membrane via a protein complex termed SNARE, which contains membrane attached (t-SNARE) and vesicle attached (v-SNARE) proteins. The fusion occurs in response to a calcium inflow, and the vesicle protein Synaptotagmin (Syt) serves as a calcium sensor. A cytosolic protein Complexin (Cpx) interacts with the SNARE complex, restricting the spontaneous fusion. Although molecular interactions of these proteins have been extensively studied, it is still debated how Syt dynamically interacts with the SNARE protein complex, Cpx, and lipid bilayers to trigger lipid merging. To elucidate these mechanisms, we combined molecular dynamics (MD) simulations with molecular biology and genetic approaches in Drosophila. Basing on MD simulations, we created a model of the protein fusion machinery wherein Cpx dynamically interacts with v-SNARE, preventing full SNARE assembly. Our MD simulations also elucidated how Syt interacts with lipid bilayers, causing lipid bulging that may precede the formation of the stalk and the fusion pore opening. Finally, our simulations predicted direct interactions of Syt with the SNARE-bound Cpx. The developed molecular model enabled us to predict new mutations in v-SNARE and Cpx that alter the fusion process. To test these predictions, we generated Drosophila lines with single point mutations and investigated how these mutations affect the kinetics of transmitter release. The results of these experiments suggest that our model creates the basis for systematic approach to manipulating the fusion machinery based on theoretical predictions derived from MD simulations.
References:
- Feliciano P., H. Matos, R. Andrade, and M. Bykhovskaia. 2017. Synapsin II regulation of GABAergic synaptic transmission is dependent on interneuron subtype. J. Neurosci. doi: 10.1523/JNEUROSCI.0844-16.2016.
- Sabeva N., Cho. R., Vasin, A. Gonzalez, J.T. Littleton, and M. Bykhovskaia. (2016) Complexin Mutants Reveal Partial Segregation Between Recycling Pathways that Drive Evoked and Spontaneous Neurotransmission J Neurosci 3:383-396.
- Vasin, A., Volfson, D., Littleton, J. T., and Bykhovskaia, M. (2016) Interaction of the Complexin Accessory Helix with Synaptobrevin Regulates Spontaneous Fusion, Biophys J 111, 1954-1964.
- Byhhovskaia M. 2015. Calcium binding promotes conformation flexibility of neuronal calcium sensor Synaptotagmin 1. Biophys. J. 108:22507-20.
- Fourtoul, N., Singh P., Hui C.Y., Bykhovskaia M., and A. Jagota. 2015. Coarse-Grained Model of the Snare Complex Determines the Number of Snares Required for Docking. Biophys. J. 108:2258-69.
Senyon Teddy Choe
University of California San Diego, USA
Title: Designer Biologics: BMP Chimeras and their clinical potential

Biography:
Senyon Teddy Choe, is a Professor of Biology and the founding director of Drug Discovery Collaboratory at UCSD. After his Ph.D study in Biophysics and Medical Physics at Univ. California, Berkeley. He joined the Salk Institute in 1993 as the founding faculty member of the Institute’s new Structural Biology Laboratory, and remained so through 2015. His research group has focused on understanding how cells talk to each other. An extension of these works explores designing synthetic biologics to modulate stem cells and sick cells directly. His major honors include election in 1999 to the Fellow of American Association for the Advancement of Science. He recently founded joint Center for Biosciences to translate discovery to medical applications to focus on protein engineering and developing new stem cell therapy. He currently leads Mogam Institute for Biomedical Research in Korea aiming for biologics discovery in the areas of infectious diseases and cancer.
Abstract:
Discovery of new biologics presents new challenges and opportunities and revenues for biologics are rapidly growing in biopharmaceutical industry. We exploited our detailed structural knowledge of three-dimensional structures of BMPs, their receptors, and their antagonists to engineer synthetic BMP ligands. By use of a novel protein engineering strategy that we termed RASCH (Random Assembly of Segmental Chimera and Heteromers), we have set out to design a synthetic biologics (synbiologics®) for bone and cartilage therapy. In the case of bone fusion, we have used Activin and BMP-2, which are the members of TGF-beta superfamily, to create AB204 (Allendorph et al., 2011). AB204 is a synthetic 50:50 chimera of the two ligands, which indeed shows highly effective bone-forming capability (Yoon et al., 2014). In the case of cartilage, we have used Activin and BMP-6 to create AB604, again a 50:50 chimera of the two. AB604 shows all the properties of super BMP6, surpassing the functional characteristics of natural BMP6.
This approach promises to be a very powerful way to harness different biological functionalities of natural ligands to merge into one synthetic designer molecule such that it goes beyond what Mother Nature could provide a new means to meet various unmet clinical needs.
References:
- Allendorph, G., Read, J.D., Kawakami, Y., Kelber, J.A., Isaacs, M.J., Choe, S. (2011) Designer TGF-beta superfamily ligands with diversified functionality. PLoS One, 6, e26402.
- Kwiatkowski, W., Gray, P.C., Choe, S. (2014) Engineering TGF-beta superfamily ligands for clinical applications. Trends Pharmacol. Sci., 35:648-57.
- Yoon, B., Esquivies, L., Ahn, C., Gray, P. C., Ye, S.-K., Kwiatkowski, W., Choe, S. (2014) An Activin A/BMP2 chimera displays osteogenic activity superior to that of BMP2. J. Bone and Mineral Research. Online April 1, 29:1950-9
- Yoon, B., Lee, J.H., Na, K., Cho, J., Choe,S. (2015) The toxicological evaluation of repetitive 2- and 4-week intravenous injection of Activin A/BMP-2 chimera (AB204) into rats. Regulatory Toxicology and Pharmacology, 73, 1-8.
- Yoon, B., Lee, J.H., Na, K., Ahn, C., Cho, J., Lee, S.-H., Ahn, H.C., Choi, J., Oh, H., Kim, B.M., Choe, S. (2016) The effects of a single intravenous injection of novel Activin A/BMP-2 chimera (AB204) on toxicity and the respiratory and central nervous systems. Drug and Chemical Toxicology, 39(3):284-9.
John B. Bruning
The University of Adelaide, Australia
Title: Structural mechanism of partial agonists and antagonists of PPARgamma for use as antidiabetics

Biography:
I received by BSc from Texas A&M University in 1997. I began crystallography in the laboratory of Yousif Shamoo at Rice University. During my graduate studies I worked on the structural mechanism of the human sliding clamp and its interactions with DNA replication proteins. I received my PhD in 2005. I completed 2 successful post-docs. The first was at the Scripps Research Institute from 2005-2007 working on structural studies of nuclear receptors including PPAR, RXR, ER, and TR. My second post-doc was with Jim Sacchettini in the Houston Medical centre and I was a part of the TB structural genomics consortium. I received my first faculty position at the University of Adelaide in 2012 as a lecturer. I was tenured in 2015 and promoted to Senior Lecturer in 2016. Do to my continued collaboration with Scripps, I was also appointed adjunct professor of the Scripps Research Institute in 2016.
Abstract:
Synthetic full agonists of PPARγ have been prescribed for the treatment of diabetes due to their ability to regulate glucose homeostasis and insulin sensitization. While the use of full agonists of PPARγ has been hampered due to severe side effects, partial agonists and antagonists have shown promise due to their decreased incidence of such side effects in preclinical models. No kinetic information has been forthcoming in regard to the mechanism of full versus partial agonism of PPARγ to date and little structural and dynamic information is available which can shed light on the mechanistic difference between full and partial agonists as well as antagonists. We have used X-ray crystallography, cellular assays, Hydrogen Deuterium Exchange (HDX), and Surface Plasmon Resonance (SPR) to probe the mechanism of several PPARγ partial agonists and antagonists. Our findings demonstrate that not only do partial agonists and antagonists act through distinct transcriptional mechanisms, they also demonstrate differences in structure, dynamics, and kinetics as compared to full agonists.
References:
- “X-ray Crystal Structure of Rivoglitazone bound to PPARγ and PPAR Subtype Selectivity of TZDs.” Rajapaksha H, Bhatia H, Wegener K, Petrovsky N, Bruning JB. Biochim Biophys Acta. (2017 May 9).
- “Structure-Activity Relationship of 2,4-Dichloro-N-(3,5-dichloro-4-(quinolin-3-yloxy)phenyl)benzenesulfonamide (INT131) Analogs for PPARγ-Targeted Antidiabetics.” Frkic RL, He Y, Rodriguez BB, Chang MR, Kuruvilla D, Ciesla A, Abell AD, Kamenecka TM, Griffin PR, Bruning JB. J Med Chem. (2017 May 22).
- “PPARG Post-translational Modifications Regulate Bone Formation and Bone Resorption.” Stechschulte LA, Czernik PJ, Rotter ZC, Tausif FN, Corzo CA, Marciano DP, Asteian A, Zheng J, Bruning JB, Kamenecka TM, Rosen CJ, Griffin PR, Lecka-Czernik B. EBioMedicine. (2016 Aug;10:174-84).
- “SR2067 Reveals a Unique Kinetic and Structural Signature for PPARγ Partial Agonism.” van Marrewijk LM, Polyak SW, Hijnen M, Kuruvilla D, Chang MR, Shin Y, Kamenecka TM, Griffin PR, Bruning JB. ACS Chem Biol. (2016 Jan 15);11(1):273-83.
- “Structural mechanism for signal transduction in RXR nuclear receptor heterodimers.” Kojetin DJ, Matta-Camacho E, Hughes TS, Srinivasan S, Nwachukwu JC, Cavett V, Nowak J, Chalmers MJ, Marciano DP, Kamenecka TM, Shulman AI, Rance M, Griffin PR, Bruning JB, Nettles KW. Nat Commun. (2015 Aug 20);6:8013.
Luba Tchertanov
CMLA ENS Cachan, France
Title: How Conformational Dynamics Descriptors may help in remodeling of allosteric Regulation in Proteins
Biography:
Luba Tchertanov, Research Director at CNRS-France, leader of the Bioinformatics, Molecular Dynamics and Modeling (BiMoDyM) team in Centre Mathématiques et leurs Applications (CMLA-CNRS) at the Ecole Normale Supérieure (ENS) de Cachan. She has multidisciplinary high-level skills, with extensive experience in structural biology, molecular modelling and numerical simulation (more than 100 papers in peer-reviewed journals). She coordinated or contributed as team-leader to different research projects (CEE, ANR, Fondation de France, OSEO, SIDACTION) and industrial contracts (LIPHATECH, the SERVIER Institute, the Pierre FABRE Laboratory, UNILIVER).
The research topics are focused on exploration of protein structure–dynamics–function relations. In particular, L. Tchertanov is working at the mechanisms of the receptors activation, the mechanisms of resistance to inhibitors, the conformational plasticity and dynamics of inter-molecular interactions and molecular recognition. She is most specifically interested in description of allosteric regulation at an atomistic level. Important part of research is dedicated to the development of new methodology and computing tools for description of proteins dynamics.
Abstract:
Allostery controls nearly all biological processes, and it has been declared Monod to be “the second secrete of life” after the genome. This universal phenomenon in nature represents a target response on a perturbation (e.g. a ligand binding) leading to a functional change at the target through alteration of the structure or dynamics. Such an event can be described in terms of a large-scale transmission of information between residues. This concept is the cornerstone of our method MONETA that delivers descriptor encoding of the communication network in a protein [1]. Using MONETA, we described the allosteric regulation of several proteins involved in cell signalling. Studying the receptors tyrosine kinases (RTKs), KIT and CSF-1R, and their numerous clinically-relevant mutants, we showed that the allosteric communications between the major regulating fragments in the native proteins were disrupted by the gain-of-function mutations [2-4]. The diverging impact of equivalent mutations on communication in homologue RTKs permits us to distinguish between the mutation-induced effects that lead to the constitutive activation of KIT and the mutation-induced effects promoted the resistance in CSF-1R. In STAT5s, RTK downstream signalling proteins, we showed the sequence-dependent asymmetry in the STAT5s’ communications and their different responses to phosphorylation [5]. Our recent study provided a fascinating illustration of how the binding of agonist ligands controls intrinsic conformational dynamics in human NMDA receptors that stabilize the channel opening. The allosteric binding sites, which were identified by a pockets search at the proteins surface adjacent to the communication pathway (Figure 1), may constitute valid targets for the development of inhibitors able to modulate the function-related communication properties of a protein. Such communication-inspired and communication-targeted modulation may selectively block several activation or post-transduction processes. Our work opens the way to novel and rational strategies for the definition of targets, and the development of efficient target-specific inhibitors.
References:
- Allain A, Chauvot de Beauchêne I, Langenfeld F, Guarracino Y, Laine E, Tchertanov L. (2014). Allosteric Pathway Identification through Network Analysis from Molecular Dynamics Simulations to Interactive 2D and 3D Graphs. Faraday Disc., 169, 1-18.
- Chauvot de Beauchêne I, Alain A, Panel N, Laine E, Trouvé A, Dubreuil P. and Tchertanov L. (2014). Oncogenic mutations of KIT receptor differentially modulate tyrosine kinase activity and drug susceptibility. PLOS Comput. Biol.;10(7):e1003749.
- Da Silva Figueiredo Celestino Gomes P, Chauvot de Beauchêne I, Panel N, Pascutti P, Solary E, Tchertanov L. Insight on Mutation-Induced Resistance from Molecular Dynamics Simulations of Wild-type and mutated CSF-1R and KIT. (2016) PLoS ONE.11(7):e0160165.
- Chauvot de Beauchêne I, Tchertanov L. (2016). How Missense Mutations in Receptors Tyrosine Kinases impact Constitutive Activity and alternate Drug Sensitivity: Insights from Molecular Dynamics Simulations. Invited Research highlight for Receptors & Clinical Investigation. Vol.3, e1372.
- Langenfeld F, Guarracino Y, Arock M, Trouvé A,Tchertanov L. (2015). How intrinsic molecular dynamics controls intramolecular communication in Signal Transducers and Activators of Transcription Factor STAT5. PLOS ONE. Dec 30;10(12):e0145142.
Sangwook Wu
Pukyong National University, South Korea
Title: Study on the conformational transition between the alternative and collapsed form of prethrombin-2: targeted molecular dynamics and free energy sampling
Biography:
Sangwook Wu received his B.A. degree in Biochemistry from Yonsei University (Korea) in 1990. After working as a Scientist at Samsung Display Devices (1995-1999), he obtained his Ph.D. in theoretical condensed matter physics from Iowa State University (1999-2005). He joined the computational biophysics lab at UNC-Chapel Hill (Dr. Lee Pedersen) as postdoctoral research associate (2005-2014). From 2014 to the present, he has been a faculty member at Pukyong National University in Korea. His research interests are in the areas of computational dynamics of biological macromolecules.
Abstract:
The alternative and collapsed forms of prethrombin-2 are revealed by X-ray crystallogrphy. We analyzed the conformational transition from the alternative to the collapsed form employing targeted molecular dynamics simulation and 2-dimensional free energy landscape using WHAM method. Some hydrophobic residues (W60d, W148, W215, and F227) show a significant difference between the two conformations in the conformational transition process. We show that the four hydrophobic residues undergo concerted movement from dimer to trimer transition via tetramer state in the conformational change from the alternative to the collapsed form. Also, we reveal that the concerted movement of the four hydrophobic residues is controlled by movement of specific loop regions behind. In this study, we discuss the difference between the transition path generated by the targeted Mplease let m eD simulation and the transition path with minimum Boltzmann weighting on the two-dimensional free energy surface (FES).
References:
- S. Wu (2016) Loop-driven conformational transition between the alternative and collapsed form of prethrombin-2: Targeted Molecular Dynamics study, Journal of Biomolecular Structure and Dynamics, DOI:10.1080/07391102.2015.1134347.
- S. Wu, C. J. Lee, and L. G. Pedersen (2014) Analysis on long-range residue–residue communication using molecular dynamics, Proteins: Structure, Function and Bioinformatics, 82: 2896 .
- S. Wu, W. A. Beard, L. G. Pedersen, and S. Wilson (2014) Structural comparison of DNA polymerase architecture suggest a nucleotide gateway to the polymerase active site”, Chemical Review, 114: 2759.
- S. Wu, J. Tie, D. W. Stafford, and L. G. Pedersen (2014) Membrane topology for human vitamin K epoxide reductase (VKOR), J Thromb Haemost, 12:112.
- S. Wu, S. Liu, S. Sim, L. G. Pedersen (2012) Weak antiferromagnetic coupling via a superexchange interaction between Mn (II)–Mn (II) ions: A QM/MM study of the active site of human cytosolic X-propyl aminopeptidase P”, Journal of Physical Chemistry Letter, 3: 2293.
Ramakrishna Vadrevu
Birla Institute of Technology and Science, India
Title: Structural and Molecular Dynamics Analysis of the super secondary motifs from TIM barrel proteins: Implications for folding and engineering of foldable building blocks for the assembly of TIMs

Biography:
Ramakrishna Vadrevu received his Masters degree in Physical Chemistry and his Ph.D. degree in Biophysical Chemistry. He spent few years at the Pennyslvania State University and later at University of Massachusetts Medical School as a post doctoral fellow with Prof. C. Robert Mathews. Since 2008 he has been a faculty at the Birla Institute of Technology and Science-Pilani, Hyderabad Campus in the department of Biological Sciences.
His research focuses on understanding the role cellular environment on protein stability and folding. His research interests also include protein design and engineering, amyloid material and its applications.
Abstract:
Statement of the Problem: The acquired complex three-dimensional structure of proteins is a culmination of simple structural fragments like a-a, b-b, a-b and b-a units. Thus, tertiary structures can be seen as a combination of basic building block motifs implying that all complex protein structures have evolved from the assembly of small independently folding super secondary structures. The TIM barrel proteins are made up of a regular repeating bab motif resulting in the strands and helices in an alternating repetitive pattern. Experimental and theoretical studies have revealed that bab unit acts as a minimal unit of stability. The success of designing super secondary motifs that fold in isolation underscores the prospects of designing and or identification of independently folding motifs from the existing protein structures. However, intriguingly, naturally occurring bab sequences from proteins that fold independently have not been identified. In our attempts we addressed the finding of ‘needles in hay stick’ scenario by an exhaustive sequence and structural space search of the bab units from the TIM barrels. Methodology & Theoretical Orientation: The search approach implemented in this work considered features such as alpha helical propensity, loop length, loop dynamics, residue preferences in loops, long range side chain main chain interactions etc., to shortlist bab units with strong propensity to fold in isolation. The prospective bab candidates thus shortlisted from the TIM barrels have been further subjected to structure forming tendency employing a combination of Monte Carlo and Molecular dynamics simulations to assess their foldability and stability. Conclusion & Significance: The prediction of some independently folding bab candidates from TIMs are enabling us to experimentally asses their folding and stability. The identification and analysis of independently folding bab units that exist naturally will not only provide substantial information on nature’s design strategies and evolution of protein conformations but also help to design/engineer novel proteins.
References:
- Grishin NV (2001). Fold change in evolution of protein structures. J Struct Biol. 134:167-85.
- Höcker B (2014). Design of proteins from smaller fragments-learning from evolution. Curr Opin Struct Biol. 27:56-62.
- Friedland GD, Kortemme T (2010). Designing ensembles in conformational and sequence space to characterize and engineer proteins. Curr Opin Struct Biol. 20:377-384.
- Riechmann L , Winter G (2006). Early protein evolution: building domains from ligand-binding polypeptide segments. J. Mol. Biol. 353:460-468
- Yang X, Kathuria SV, Vadrevu R, Matthews CR. (2009). Beta alpha- hairpin clamps brace beta alpha beta modules and can make substantive contributions to the stability of TIM barrel proteins. PLoS One. 4(9):e7179
- Special Session on Structural Biology of Biomembranes
Session Introduction
Qiu-Xing Jiang
University of Florida, USA
Title: Structural basis for the lipid-dependent gating of a Kv channel

Biography:
Qiu-Xing Jiang is currently heading the Laboratory of Molecular Biophysics and Cell Physiology in the Department of Microbiology and Cell Science in the Institute of Food and Agricultural Sciences of University of Florida (UF), and he is serving part-time (20%) as the faculty director of Electron Microscopy at the Institute of Cross-disciplinary Biotechnology Research (ICBR) at UF. His lab is working on lipid-dependent gating effects and on a newly discovered anion conductance in ithe regulated secretory pathways. Dr. Jiang obtained his PhD in 2002 from the Department of Cellular and Molecular Physiology at Yale University School of Medicine, where he started his work in cryo-electron microscopy (cryoEM) in 1999 with Dr. Fred Sigworth. He initiated the implementation of spherically constrained reconstruction (SCR) by imaging membrane proteins in small vesicles. After a short postdoctoral training at Yale, Dr. Jiang finished his postdoctoral training in structural biology with Dr. Roderick Mackinnon in 2007 before taking an Assistant Professorship position at UT Southwestern Medical Center at Dallas, Texas. During the time at Dallas, Dr. Jiang worked on the development of chemically functionalized carbon films for cryoEM imaging, and made new discoveries on the filament-based signal amplification in the innate immune response against RNA viruses. He studied membrane-induced pore formation by human C-type Lectin and the VopQ proteins, and discovered the mechanism for lipid-dependent gating of Kv channels. He has published 24 papers in international journals. He is the recipient of the NIH EUREKA award in 2009, the AHA National Innovative Award in 2012, and the Junior Faculty travel award from GRC Molecular and Cell Biology of Lipids in 2011.
Abstract:
Human cell membranes are made of both phospholipids and nonphospholipids. The nonphospholipids, such as cholesterol (CHOL), have no phosphate groups in their headgroup regions and are quite abundant in cell membranes. Mainly due to technical difficulties, quantitative study of possible effects of nonphospholipids on voltage-gated ion channels has been very challenging. Our prior studies have achieved three major developments: 1) a working hypothesis of lipid-dependent gating based on nonphospholipids stabilizing the voltage sensor domain (VSD) of the KvAP channel in the resting (“down”) conformation; 2) a novel bead-supported unilamellar membrane (bSUM) system and a new method to stabilize the KvAP channel in the resting state; and 3) chemically functionalized carbon (ChemiC) films for cryoEM imaging of low abundance complexes by high-affinity selection or of small macromolecular complexes (100-200 kDa) by keeping vitrified ice thinner than usual. The general idea for lipid dependent gating is that the annular lipids around a Kv channel change their arrangements in accompany with the conformational changes of the voltage-sensor domains. Our technical development made it feasible to study the CHOL-dependent gating effects on Kv channels. We studied the CHOL-dependent gating effects on Kv channels in bSUMs. Because almost all known lipid metabolic defects result from dysregulated homeostasis of nonphospholipids, our studies in animal models carrying CHOL metabolic defects will provide the first test of lipid-dependent gating in an in vivo physiological setting. Secondly, we apply our ChemiC method to cryoEM study of the 120 kDa KvAP in both an inactivated and a peptide-stabilized “down” state. The peptides selected from the nonphospholipid-stabilized down state have been showed to recognize the voltage sensors in the right conformation and keep the channels in the right conformation. Our results will reveal the structural basis for the nonphospholipid-induced conformational changes in Kv channels, and unveil connections to the lipid-metabolic defects in humans.
References:
- Hui Zheng, Weiran Liu, Lingyan Anderson, and Qiu-Xing Jiang. Lipid-dependent gating of a voltage-gated ion channel. Nature Comm., 2:250 doi:10.1038/ncomms1254, 2011.
- Marc Llaguno, Hui Xu, Liang Shi, Nian Huang, Hong Zhang, Qinghua Liu, and Qiu-Xing Jiang. (2014) Chemically functionalized nanometer-thick carbon films for single molecule imaging. J. Struct. Biol. 185(3):405-17.
- Hui Zheng, Sungsoo Lee, Marc Llaguno, and Qiu-Xing Jiang. (2016). bSUM: A bead-supported unilamellar membrane system facilitating unidirectional insertion of membrane proteins into giant vesicles. J Gen Physiol. 147(1):77-93.
- Hui Xu, Xiaojing He, Hui Zheng, Lily J. Huang, Fajian Hou, Zhiheng Yu, M. Jason de la Cruz, Brian Borkowski, Xuewu Zhang*, Zhijian J. Chen* and Qiu-Xing Jiang*. (2014) Structural basis for the prion-like MAVS filaments in antiviral innate immunity. eLife. 3:e01489. doi: 10.7554/eLife.01489.
- Sohini Mukherjee, Hui Zheng, Mehabaw Derebe, Keith Callenberg, Carrie L. Partch, Darcy Rollins, Daniel C. Propheter, Josep Rizo, Michael Grabe, Qiu-Xing Jiang*, and Lora V. Hooper*. (2014) Antibacterial membrane attach by a pore-forming C-type lectin. Nature, 505(7481): 103-7. (*co-senior authors).
John E. Baenziger
University of Ottawa, Canada
Title: Mechanisms underlying lipid-sensing by the nicotinic acetylcholine receptor in both normal and diseased states

Biography:
John Baenziger is a professor of Biochemistry at the University of Ottawa in Ottawa, Canada. His research is focused on understanding the mechanisms by which lipids influence nicotinic acetylcholine receptor structure and function in both normal and diseased states, with increasing focus on how lipid-nAChR interactions participate in congenital myasthenic syndromes. Dr. Baenziger has served on the editorial board of the Journal of Biological Chemistry. He is the President of the Biophysical Society of Canada and is Treasurer-elect of the International Union of Pure and Applied Biophysics.
Abstract:
The neuromuscular nicotinic acetylcholine receptor (nAChR) is the prototypic member of the pentameric ligand-gated ion channel (pLGIC) superfamily, a superfamily of neurotransmitter receptors that plays a central role in information processing in the brain. It is well documented that nAChR function is exquisitely sensitive to its lipid environment. Lipids influence function by both conformational selection and kinetic mechanisms – they stabilize different proportions of activatable versus non-activatable conformations, and influence the rates of transitions between the different states. In the absence of activing cholesterol and anionic lipids, the nAChR adopts a conformation where agonist binding is uncoupled from channel gating. Lipids likely influence the “coupling” of binding and gating via the lipid-exposed transmembrane a-helix, M4. M4 in the neuromuscular nAChR is also the site of both point and truncation mutations that alter expression and/or function leading to congenital myasthenic syndromes. In this seminar, I will focus on the mechanisms by which the peripheral M4 transmembrane a-helix modulates pLGIC function. The M4 C terminus extends beyond the bilayer to interact with key structures that link the agonist binding to the transmembrane gate – referred to here as the coupling interface. We hypothesized that interactions between M4 and the coupling interface are essential to pLGIC function. We show here that such interactions are essential to function in some pLGICs and do participate in lipid-sensing. In the neuromuscular nAChR, however, such interactions between M4 and the coupling interface are less important. Instead, M4 influences function via a cluster of polar residues located in the core of the transmembrane domain near the center of the lipid bilayer. Altered M4 structure leads to changes in the energetic coupling between these polar residues, with the changes coupling ultimately propagating to both the gating helix, M2, and the aforementioned coupling interface. Here, we map out the conformational pathway that leads from the lipid-exposed surface of M4 to the channel gate, and thus illustrate how M4 “allosterically” modulates channel function.

Figure 1: Some allosteric modulators, including lipids, act via the lipid-exposed M4 a-helix of the nAChR. We elucidate the allosteric pathway by which this peripheral structure influences channel gating.
References:
- Baenziger JE et al (2017) The role of cholesterol in the activation of nicotinic acetylcholine receptors. Current Topics in Membranes article in press
- Therien JPD, Baenziger JE (2017) Pentameric ligand-gated ion channels exhibit distinct transmembrane domain archetypes for folding/expression and function. Scientific Reports 7:450:1-14.
- Carswell CL et al (2015) Role of the fourth transmembrane a‑helix in the allosteric modulation of pentameric ligand-gated ion channels. Structure 23:1655-1664.
- Baenziger JE et al (2015) Nicotinic acetylcholine receptor-lipid interactions: mechanistic insight and biological function. BBA-Biomembranes 1848:1806-1817.
- daCosta CJB et al (2013) A novel mechanism for activating uncoupled nicotinic acetylcholine receptors. Nat Chem Biol 9:701-707
Biography:
Carsten Mim has a longstanding interest in membrane and membrane-associated proteins throughout his career. As an experienced electrophysiologist, he characterized the glutamate transporter EAAT3 and EAAT4. The kinetics of EAAT4 differ from other glutamate transporters, by a voltage sensitive step that slows the turnover rate at hyperpolarized membrane potentials. Further, recorded transient and steady state currents at different temperatures showed that the binding of glutamate is enthalpy-driven unlike the binding of Na+. To visualize membrane:protein complexes, he turned to electron microscopy. His work on the Bin/Amphyphysin/Rvs domain (BAR) protein endophilin in complex with the bilayer resulted in the unexpected discovery that the stability and dynamics of endophilin scaffolds entirely depend on non-specific interactions between amphipathic helices in the bilayer. His findings also provided a first structurally motivated hypothesis how BAR-scaffolds selectively recruit downstream interaction partners through a steric selection mechanism.
Abstract:
Statement of the Problem: The cell bends membranes to generate membrane structures, like the t-tubules in muscles. Bin1/Amphiphysin/Rvs domain (BAR) proteins are part of the membrane bending machinery and are found in widespread phenomena like endocytosis or cell motility. BAR domain proteins can assemble spontaneously in vitro as well as in vivo. Which factors regulate the assembly? The membrane tension is a well studied regulator. In contrast, the role of the membrane composition, as an initiator of membrane bending, is poorly understood. Methodology & Theoretical Orientation: For this study we collected electron micrographs to document the membrane bending activity of the BAR protein Bin1. We probed the electrostatic interactions between Bin1 and the membrane by changing the surface charge of the membrane, the ionic strength of the assay and using disease relevant mutants, where a positive charge (K35N) and a negative charge (D151N) are eliminated. The electrostatic interactions between Bin1 and artificial membranes were evaluated by liposome sedimentation. To test how the findings translate into living cells, we assayed the phenotype of membrane bending-deficient Bin1 mutants in cells that have elevated or reduced levels of negatively charged lipids. Findings: Our simple, artificial system could reproduce the complex membrane topology present in muscle cells. We focused on the two mutants. We found that in stringent conditions for membrane bending (high ionic strength, low membrane charge) the mutants showed disproportional lower bending activity. These finding were confirmed in vivo. We were able to rescue to mutant phenotype by increasing the membrane surface charge. Conversely, we induced a mutant phenotype in wt Bin1 by lowering the membrane surface charge. Conclusion & Significance: We established the membrane charge as a novel regulator of membrane tubulation. We speculate that rapid phosphorylation and dephosphorylation of phosphoinositols can act as a switch for induction of membrane bending.

References:
- Higher-order assemblies of BAR domain proteins for shaping membranes. Suetsugu S. Microscopy (Oxf). 2016 Feb 15
- Membrane tension and peripheral protein density mediate membrane shape transitions. Shi Z, Baumgart T., Nat Commun. 2015 Jan 8;6:5974
- Structural basis of membrane bending by the N-BAR protein endophilin. Mim C, Cui H, Gawronski-Salerno JA, Frost A, Lyman E, Voth GA, Unger VM., Cell 2012 Mar 30;149(1):137-45
- Protein-mediated transformation of lipid vesicles into tubular networks. Simunovic M, Mim C, Marlovits TC, Resch G, Unger VM, Voth GA. Biophys J. 2013 Aug 6;105(3):711-9

Biography:
Volodymyr Korkhov is an assistant professor at the Institute of Biochemistry and Paul Scherrer Institute (PSI, Villigen). Prof. Korkhov has been studying various aspects of membrane protein biology throughout his career. As a PhD student at the Institute of Pharmacology, Vienna Medical University, he studied oligomerization of neurotransmitter transporters. He continued research of multidrug and ABC transporters during his postdoctoral training periods at MRC Laboratory of Molecular Biology (Cambridge, UK) and ETH Zurich, respectively. His work on ABC transporter for vitamin B12 from Escherichia coli, BtuCDF, led to a proposal of a complete structure-based mechanism of type II ABC importers. From April 2014, Prof. Korkhov has been leading an independent research group, supported by an SNF Professorship. The overarching topic of research in Prof. Korkhov’s group is structure and molecular mechanisms of membrane protein complexes involved in signal transduction.
Abstract:
- Track 8: Drug Designing