Day 2 :
Keynote Forum
Timothy A. Cross
Florida State Univ. & National High Magnetic Field Lab, USA
Keynote: Membrane Protein Structure, Dynamics & Function: Oriented Sample and Magic Angle Sample Spinning Solid State NMR

Biography:
Timothy Cross has more that 30 years of experience characterizing membrane proteins in lipid bilayer environments using solid state NMR of liquid crystalline bilayer preparations of peptides and proteins. This has brought to light a fundamental understanding of membrane protein biophysics that has led to detailed functional characterizations of membrane channels – in particular the gramicidin monovalent cation channel and the influenza A M2 proton channel. In both systems the unique features of the membrane environment play crucial roles in the functional mechanisms and kinetics of ion conductance. Is this understanding of the influence of membrane and lipid environments through the use of solid state NMR that has driven his research at the frontier of membrane protein biophysics.
Abstract:
Statement of the Problem: Unlike water soluble proteins that have a relatively homogeneous environment, membrane proteins exist in a dramatically heterogeneous environment. The result is protein structure that is stabilized by a different balance of molecular interactions for the membrane embedded portion of the protein compared to the water soluble or membrane interfacial regions of the protein. The result is a need to model the membrane environment as closely as possible to that of the native environment for structural, dynamic and functional characterizations. Methodology: Biological solid state NMR provides a unique opportunity to model the membrane environment with liquid crystalline lipid bilayers and a wide variety of lipids. The samples can be prepared either as liposomes for magic angle sample spinning (MAS) or as uniformly oriented samples (OS) for the spectroscopy. The former provides solution like spectra for both distance and isotropic chemical shift restraints, while oriented samples provide absolute restraints that restrain the atomic sites in the protein structure to the bilayer normal. In addition to structural restraints it is possible to characterize the protein’s dynamics and kinetic rates. Findings, Conclusion & Significance: The structure, dynamics and kinetics associated with the M2 proton channel from influenza A have been characterized yielding a unique mechanism for proton transport by this important drug target. In addition, the cholesterol binding to M2 has been found to stabilize the amphipathic helix in the lipid interface an essential feature for this protein’s functional role in viral budding. Recent structural studies of the CrgA protein from Mycobacterium tuberculosis have characterized a dimeric structure stabilized primarily by intermolecular b-sheet in the membrane interfacial region. The protein is part of the cell division apparatus and appears to play a role in recruiting multiple proteins to the divisome, potentially through its transmembrane domain.
References:
1. Zhou, H.-X. & Cross, T.A. (2013) “Influences of Membrane Mimetic Environments on Membrane Protein Structures” Annual Reviews of Biophysics 42:361-392.
2. Sharma, M., Yi, M., Dong, H., Qin, H., Petersen, E., Busath, D.D., Zhou, H.-X. & Cross, T.A. (2010) “Insight into the Mechanism of the Influenza A Proton Channel from a Structure in a Lipid Bilayer” Science 330:509-512.
3. Murray, D., Das, N., Cross, T.A. (2013) “Solid State NMR Strategy for Characterizing Native Membrane Protein Structures” Accounts of Chemical Research 46:2172-2181.
4. Miao, Y., Fu, R., Zhou, H.-X. & Cross, T.A. (2015) Dynamic, Short Hydrogen Bonds in Histidine Tetrad of Full-Length M2 H+ Channel Reveals Tetrameric Structural Heterogeneity and Functional Mechanism” Structure 23:2300-2308.
5. Das, N., Dai, J., Hung, I., Rajagopalan, M., Zhou, H.-X. & Cross, T.A. (2015) “Structure of CrgA, a Cell Division Structural and Regulatory Protein from Mycobacterium tuberculosis in Lipid Bilayers” Proc. Natl. Acad. Sci. 112:E119-E126.
Keynote Forum
Wladek Minor
University of Virginia, USA
Keynote: Reproducibility in Biomedical Sciences - Big Data Perspective

Biography:
Wladek Minor is a Harrison Distinguished Professor of Molecular Physiology and Biological Physics at University of Virginia. He is an expert in structural biology and data mining. He is an author of over 190 papers that attracted over 37,000 of citations. His Relative Citations Ratio is above 560.He trained over 80 scientists that currently pursue career in academia, government and industry.
Abstract:
Experimental reproducibility is the cornerstone of scientific research, upon which all progress rests. The veracity of scientific publications is crucial because subsequent lines of investigation rely on previous knowledge. Several recent systematic surveys of academic results published in biomedical journals reveal that a large fraction of representative sets of studies in a variety of fields cannot be reproduced in another laboratory. Big Data approach and especially NIH Big Data to Knowledge (BD2K) program is coming to the rescue.
The goal of the presented research is to provide the biomedical community with a strategy to increase the reproducibility of reported results for a wide range of experiments by building a set of “best practices”, culled by extensive data harvesting and curation combined with experimental verification of the parameters crucial for reproducibility. Experimental verification assisted by the automatic/semi-automatic harvesting of data from laboratory equipment into the already developed sophisticated laboratory information management system (LIMS) will be presented. This data in, information out paradigm will be discussed.
References:
- Zheng H, Cooper DR, Porebski PJ, Shabalin IG, Handing KB, Minor W (2017) CheckMyMetal: a macromolecular metal-binding validation tool. Acta Cryst. D 73: 223-233
- Grabowski M, Minor W (2017) Sharing Big Data. IUCrJ 4: 3-4
- Rupp B, Wlodawer A, Minor W, Helliwell JR, Jaskolski M (2016) Correcting the record of structural publications requires joint effort of the community and journal editors. FEBS J. 283: 4452-4457
- Grabowski M, Langner KM, Cymborowski M, Porebski PJ, Sroka P, Zheng H, Cooper DR, Zimmerman MD, Elsliger MA, Burley SK, Minor W (2016) A public database of macromolecular diffraction experiments. Acta Cryst D 72: 1181-1193
- Niedzialkowska E, Gasiorowska O, Handing KB, Majorek KA, Porebski PJ, Shabalin IG, Zasadzinska E, Cymborowski M, Minor W (2016) Protein purification and crystallization artifacts: The tale usually not told. Protein Science 25: 720-33
Keynote Forum
Yuri L. Lyubchenko
University of Nebraska Medical Center, USA
Keynote: Protein-protein interaction and amyloid cascade hypothesis for Alzheimer’s disease

Biography:
Yuri L. Lyubchenko is Professor of Pharmaceutical Sciences at the University of Nebraska Medical Center, Omaha, NE, USA. His research focuses on understanding fundamental mechanisms underlying health and disease, which are key to developing new and more effective diagnostics and medications. This primarily basic research allows him not only identify new drug targets for small molecule drugs, it also leads to development of the nanotools and methods to discover novel approaches for diagnostic, treatment and disease prevention and to more rapidly determine their efficacy at the molecular level.
Abstract:
The amyloid cascade hypothesis is currently considered as the main model for a vast number of neurodegenerative diseases including Alzheimer’s, Parkinson’s, and Huntington’s diseases. Numerous studies have shown that amyloidogenic proteins are capable of spontaneous assembly into aggregates, and eventually form fibrillar structures found in amyloid or amyloidâ€like deposits. However, there is a serious complication with translating current knowledge on amyloid aggregation in vitro to understand the aggregation process in vivo. If the critical concentration for the spontaneous aggregation of Aβ peptide in vitro is in the micromolar range, physiological concentrations of Aβ are in the low nanomolar range making impossible amyloids to assemble. We have discovered a novel on-surface aggregation pathway that allows for spontaneous assembly of amyloid beta peptides at the physiological concentration range. Our combined experimental and computer modeling approaches demonstrate that the on-surface aggregation is a dynamic process, so the assembled aggregate can dissociate from the surface to the bulk solution. As a result, the dissociated oligomers can play roles of seeds for aggregation in the bulk solution, or start a neurotoxic effect such as phosphorylation of the tau protein to initiate its misfolding and aggregation. Both processes lead to neurodegeneration. Importantly, in the vast majority of cases, we found that aggregates formed on the surface are oligomers, which are considered to be the most neurotoxic amyloid aggregates. Therefore, we posit that on-surface aggregation is the mechanism by which neurotoxic amyloid aggregates are produced under physiological conditions. A change in membrane properties leading to an increase in affinity of amyloid proteins to the membrane surface facilitates the assembly of stable oligomers. The proposed model is a significant departure from the current model as it directs the development of treatments and preventions towards approaches that control the cell membranes properties and composition preventing the on-surface aggregation process.
References:
1. Lyubchenko, Y. L. (2014) Centromere chromatin: a loose grip on the nucleosome?, Nat Struct Mol Biol 21, 8.
2. Lyubchenko, Y. L., and Shlyakhtenko, L. S. (2015) Chromatin imaging with time-lapse atomic force microscopy, Methods Mol Biol 1288, 27-42.
3. Lyubchenko, Y. L., and Shlyakhtenko, L. S. (2015) Atomic Force Microscopy Imaging and Probing of Amyloid Nanoaggregates, In Handbook of Clinical Nanomedicine: From Bench to Bedside (Bawa, R., Audette, G. & Rubinstein, I., Ed.), p 1500. , Pan Stanford Publishing, Singapore. .
4. Sun, Z., Tan, H. Y., Bianco, P. R., and Lyubchenko, Y. L. (2015) Remodeling of RecG Helicase at the DNA Replication Fork by SSB Protein, Sci Rep 5, 9625.
5. Proctor, E. A., Fee, L., Tao, Y., Redler, R. L., Fay, J. M., Zhang, Y., Lv, Z., Mercer, I. P., Deshmukh, M., Lyubchenko, Y. L., and Dokholyan, N. V. (2016) Nonnative SOD1 trimer is toxic to motor neurons in a model of amyotrophic lateral sclerosis, Proc Natl Acad Sci U S A 113, 614-619.
- Track 8: Structural Biology in Complexity Arenas
Session Introduction
Charles W. Carter, Jr
University of North Carolina, USA
Title: How Does Domain Motion Contribute to Transition-State Stabilization? Combinatorial Thermodynamic Cycle Analysis of Conformational Coupling During Tryptophan Activation
Time :

Biography:
Charlie Carter is an X-ray crystallographer who studies the origin, evolution, and structural biology of aminoacyl-tRNA synthetases. His research group introduced the use of Urzymes—highly conserved structural cores that retain large fractions of the transition-state stabilization free enregies of full length enzymes—as experimental models of ancestral enzymes.
Abstract:
Enzyme mechanisms, especially those that couple NTP hydrolysis to mechanical work and information, use sophisticated dynamic networks to transduce active-site chemistry into domain motions that change binding affinities [1]. We measured and cross-validated the energetics of such networks in B. stearothermophilus Tryptophanyl-tRNA synthetase (TrpRS) using both multi-mutant and modular thermodynamic cycles [2]. Coordinated domain motions develop shear in a core packing motif conserved in >125 different protein superfamilies [2]. Multi-dimensional combinatorial mutagenesis showed that four side chains from this “molecular switch” move coordinately with the active-site Mg2+ ion in the transition state for amino acid activation [3]. A modular thermodynamic cycle consisting of full-length TrpRS, the Urzyme, and the Urzyme plus each of the two domains deleted in the Urzyme [4]gives similar energetics. These complementary experiments establish that catalysis and specificity in full-length TrpRS are both coupled by 5 kcal/mole to: (i) the core packing region where domain movement generates shear, and (ii) the simultaneous motion of the two domains relative to the Urzyme. Theory shows that the minimum action path algorithm estimates thermodynamically meaningful contributions of domain movement to kinetic rates [5]. Correlations between those parameters, the experimental rates, and structural variations induced in the combinatorial mutants confirm that these estimates are realistic. These results validate our previous conclusion that catalysis by Mg2+ ion is coupled to the overall domain motion (3). Computational free energy surfaces demonstrate that TrpRS catalytic domain motion itself is endergonic but is driven thermodynamically by PPi release [5,6]. Comparison of the impact of combinatorial mutagenesis on pre-steady state and steady-state rates confirm that dynamic active-site pre-organization endows TrpRS with the elusive conditionality of NTP utilization on domain motion.
References:
- Carter CW, Jr. (2017) High-Dimensional Mutant and Modular Thermodynamic Cycles, Molecular Switching, and Free Energy Transduction. Annual Review of Biophysics 46:In Press. doi:10.1146/annurev-biophys-070816-033811
- Carter CW, Jr., Chandrasekaran SN, Weinreb V, Li L, Williams T (2017) s Structural Dynamics 4:032101
- Weinreb V, Li L, Carter CW, Jr. (2012). Structure, vol 20.
- Li L, Carter CW, Jr (2013) J Biol Chem 288 (29 November):34736–34745. doi:10.1074/jbc.M113.510958
- Chandrasekaran SN, Carter CWJ (2017) Structural Dynamics 4:In press
Christophe Guyeux
Université de Bourgogne Franche-Comté, France
Title: On two ways to predict the protein folding process over a chaotic model
Time :

Biography:
Guyeux has a record of about 120 scholarly publications. Since 2010, he published 43 articles in peer-reviewed international journals (as a co-author, including the top-ranked ones in the areas of computer science and interdisciplinary applications, such as AIP Chaos, Plos One, or Clinical Infectious Diseases). He is co-author of 2 book chapters and 2 scientific monograms. He is also author of 4 software patents, 53 articles that appeared in proceeding of peer-reviewed international conferences. His topics of research encompasses bioinformatics, discrete dynamical systems, and information security.
Abstract:
In our first theoretical studies about folded self-avoiding walks, we have raised raises several questions regarding the protein structure prediction problem and the current ways to solve it. In one category of PSP software, the protein is supposed to be synthesized first as a straight line of amino acids, and then this line of a.a. is folded out until reaching a conformation that optimizes a given scoring function. The second category consider that, as the protein is already in the aqueous solvent, it does not wait the end of the synthesis to take its 3D conformation. So they consider SAWs whose number of steps increases until the number of amino acids of the targeted protein and, at each step, the current walk is stretched (one amino acid is added to the protein) in such a way that the pivot k placed in the position that optimizes the scoring function. We have proven that the two sets of possible conformations are different. So these two kinds of PSP software cannot predict the same kind of conformations.
We have proven too that the folding process G in the 2D model is chaotic according to Devaney. A consequence of this theorem is that this process is highly sensitive to its initial condition. If the 2D model can accurately describe the natural process, then this theorem implies that even a minute difference on an intermediate conformation of the protein, in forces that act in the folding process, or in the position of an atom, can lead to enormous differences in its final conformation. In particular, it seems very difficult to predict, in this 2D model, the structure of a given protein by using the knowledge of the structure of similar proteins. Let us remark that the whole 3D folding process with real torsion angles is obviously more complex. And finally, that chaos refers to our incapacity to make good prediction, it does not mean that the biological process is a random one.
References:
- Christophe Guyeux, Nicod J M, Philippe L, and Bahi J (2015) The study of unfoldable self-avoiding walks: Application to protein structure prediction software. Journal of Bioinformatics and Computational Biology 13(4): 1550009.
- Christophe Guyeux, Côté N-L, Bienia W and Bahi J (2013) Is protein folding problem really a NP-complete one? First investigations. Journal of Bioinformatics and Computational Biology 12(1): 24.
- Christophe Guyeux, Bahi J, Mazouzi K, and Philippe L (2013) Computational investigations of folded self-avoiding walks related to protein folding. Computational Biology and Chemistry 47: 246-256.
- Christophe Guyeux, Bahi J, Côté N and Salomon M (2012) Protein folding in the 2D hydrophobic-hydrophilic (HP) square lattice model is chaotic. Cognitive Computation 4(1): 98-114.

Biography:
Marta obtained Ph.D. in 2007 in nanomaterials from Cranfield University. In 2011, she joined Structural Biology group led by Alaistair Lawson at UCB. For the past five years she has been developing expertise in small molecule drug discovery using a fragment based approach. She had also been involved in the assay development for a novel antibody-enable drug discovery approach. Prior to joining UCB she was awarded a Postdoctoral Fellowship at the Institute of Food Research in Norwich to study the mono and multilayer films used to encapsulate active ingredients for a controlled and site-specific delivery within the GIT. During her time at IFR she worked on numerous projects including a commercial project for Pfizer focused on AFM imaging of proteins and polysaccharides constituting vaccines. She has also been part of the team under the leadership of Claudio Nicolletii investigating the outcome of consumption of the probiotic L. casei Shirota in subjects with Seasonal Allergic Rhinitis.
Abstract:
Protein-protein interactions (PPIs) are of critical importance in the majority of biological cellular processes including DNA repair, immune, and allergic responses. Despite their therapeutic relevance, PPIs are intrinsically challenging targets due to complexity of interactions, assay tractability and the lack of well-defined binding pockets at the interacting surfaces.
In the quest for small molecule drug candidates targeting PPIs, there have been a number of different approaches adopted, which include: use of existing lead or drugs, natural products, high-throughput screening and more recently established powerful fragment-based drug discovery.
At UCB we have integrated the fragment-based methodology with biological and structural information obtained from antibody-validated protein targets, to develop specific small molecule inhibitors of PPIs. An ensemble of biophysical methods (i.e. SPR, ITC, FRET, MS and ligand-based NMR), corroborated by functional data, were employed to identify and validate fragment hits that constituted the starting point for our PPI inhibitor drug discovery programs. We have also employed antibodies as research tools to hold target proteins in biologically active conformations, aiding the discovery of new small molecules for challenging targets. By holding the target protein in biologically relevant conformations, new sites (in particular allosteric sites), which would otherwise be inaccessible, may become available for binding. The ability to capture the target protein in a specific conformation with high affinity for a significantly long time opens the possibility for a small-fragment molecule screening.
Ulf Skoglund
Okinawa Institute of Science and Technology, Japan
Title: Structure of Human IgM in complex with the Malaria protein PfEMP1

Biography:
Ulf Skoglund, born 1950, received his Ph.D. in 1969 at Stockholm University, Sweden. He was a Professor 1996 – 2009 at Karolinska Institutet, Stockholm, Sweden. Since 2010, he is a Professor in Structural Cellular Biology at Okinawa Institute of Science and Technology, Okinawa, Japan.
He has developed electron-tomographic technologies allowing for images of proteins to be generated so that e.g. X-ray structures can be fitted into the 3D densities. This technique is termed COMET (constrained maximum entropy tomography). His Unit has also developed a large-scale dynamics method that allows for quantitative calculations of molecular movements in solution. Current developments concern the mathematics and improvements of the basic 3D reconstruction principles, as well as work on reconfigurable and high performance computing. His Unit has also been actively pursuing several cell biology projects.
Abstract:
Children under the age of 5 years have huge malaria burden in endemic area. Increased death in complicated malaria is due to increased sequestration to tissues and agglutination with erythrocytes and cells of our immune system. It is known that parasites that bind to non-immune IgM cause severe malaria due to increased rosetting (agglutination). Using biochemical, parasitology and electron tomography techniques we have identified that
PfEMP1, a crescent shaped molecule interacts with human IgM through its bulky C-terminus (membrane proximal) in 1:1 and 2:1 ratio. While the bulky C terminus limits the stoichiometry of this interaction yet clusters parasite molecule PfEMP1 (P. falciparum Erythrocyte Membrane Protein-1) to mediate robust host parasite interaction. Structural analysis revealed that PfEMP1 could also preclude the activation of complement mediated lysis of parasite in spite of IgM deposition on parasitized RBC surface. We also found that IgM although not a rosetting factor enhances this interaction by increasing the strength of this interaction by at least four fold. In terms of physiological relevance, we need to understand that new born babies have high level of IgM and could be more prone to agglutination and hence more deaths due to malaria.
Matthias Buck
Case Western Reserve University, USA
Title: Solution NMR relaxation and µs Molecular Dynamics Simulations of Dynamic Protein-Protein and Protein-Membrane Complexes
Time :

Biography:
The Buck laboratory studies two receptor families responsible for cell guidance and positional maintenance (Plexins and Ephrins), both with key involvement in cardiovascular and neuronal development and disease, esp. cancer. We use a wide range of structural biology (solution NMR / x-ray crystallography) and protein biophysical tools (CD, fluorescence spectroscopy, ITC and SPR) in a problem oriented approach. Part of the laboratory also pursues computational modeling and molecular dynamics to provide additional perspective on the problems, provide new insights into the experimental data and to suggest further studies. Small GTPases and their interaction with the plexin receptor cytoplasmic domains has been a major focus of the laboratory and recently we have become very interested in protein-membrane interactions; both the transmembrane regions of the receptors as well as the transient interactions of receptor and GTPase domains with membranes
Abstract:
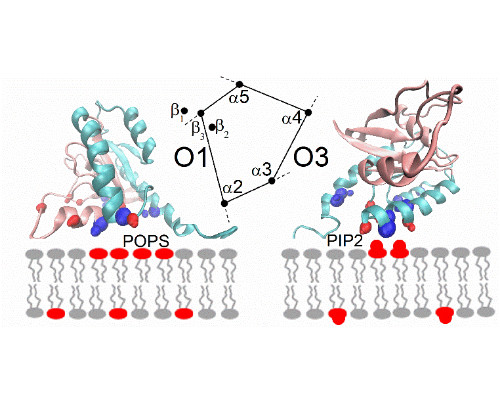
It is now recognized that protein-protein interactions in solution are often dynamic, especially if the binding affinities are only moderately strong. Dynamics originate in part from the interconversion between structures of the protein complex, e.g. one bound state that is in equilibrium with one or several alternate configurations. We determined the structure of such a complex using NMR restraints [1] and saw the transitions between different configurations in microsecond length all-atom molecular dynamics simulations [2]. Recently, we also studied the dissociation process of mutant complexes which had a weakened primary interaction interface. Those simulations suggested that there is no single dissociation pathway, but that the separation first involves transitions to binding interfaces with fewer/weaker contacts [3].
Comparison is made between experimental NMR relaxation measurements on the ps-ns as well as µs-ms timescale with the microsecond all atom simulations, also in the context of new simulations of the protein association process. The functional significance of the protein complex alternate states and their dynamics are discussed.
In a second part of the presentation, we consider a second system involving transient interactions; this time between K-Ras and the lipid bilayer of the plasma membrane [4]. Our recent simulations the full length GTPase at different membranes reveal the underlying rules of the interactions, emphasizing electrostatic contacts but also protein topology [5]. Again, simulations are compared with NMR experiments, carried out at model systems for the membrane.
References:
- Lee, HJ, Hota, P.K, Chugha, P., Miao, H., Zhang, L, Kim, SJ, Alviani, R.S, Stetzig, L., Wang, B. and Buck, M. (2012) “NMR structure of a hetero-dimeric SAM: SAM complex: Characterization and manipulation of EphA2 binding reveal new cellular functions of SHIP2”. Structure 20, 41-55.
- Zhang, L., and Buck, M. (2013) "Molecular Simulations of a Dynamic Protein Complex: Role of Salt-Bridges and Polar Interactions in Configurational Transitions" Biophys. J. 105, 2412-2417
- Zhang, L., Borthakur, S. and Buck, M. (2016) "Dissociation of a Dynamic Protein Complex studied by All Atom Molecular Simulation" Biophys. J., 110, 877-886.
- Li, Z., Cao, S., & Buck, M. (2016) “K-Ras at an Anionic Membrane: Orientation, orientation...orientation. Insights from recent simulation and experimental work”. New&Notable in Biophys. J. 110, 1033-1035.
- Li, Z. & Buck, M. (2017) “Computational Modelling Reveals Signaling Lipids Modulate the Orientation of K-Ras4A at the Membrane Reflecting Protein Topology.” Structure, revision submitted.
Beat Vögeli
University of Colorado at Denver, USA
Title: Functional protein conformation networks probed by NMR nanorulers
Time :

Biography:
Beat Vögeli has his expertise in nuclear magnetic resonance (NMR) spectroscopy of biomacromolecules. He develops methodology for the elucidation of conformation and communication networks within and between proteins and nucleic acids. He received his Ph.D. degree at the ETH Zürich in the group of Konstantin Pervushin. After a postdoctoral stay at the National Institutes of Health, Bethesda USA, in the group of Ad Bax, he returned to ETH Zürich to become Oberassistant in the group of Roland Riek and Privatdozent. He is currently an assistant professor at the University of Colorado at Denver in the Department of Biochemistry and Molecular Genetics.
Abstract:
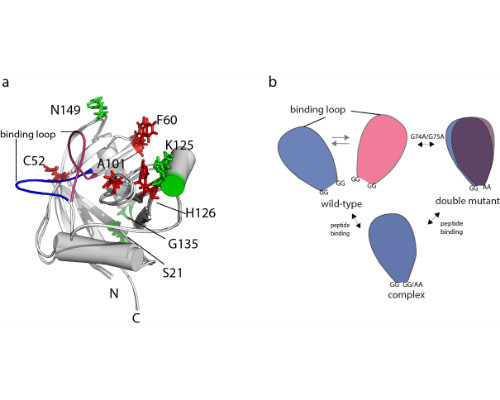
The function of a protein is tightly connected to its conformational network. Often, subtle differences distinguish interchanging states with distinct properties. One major challenge in structural biology is a sufficiently complete description of the structural landscape and the exchange dynamics between structural states at atomic resolution. We have replaced the standard NMR structure determination by an approach that generates multi-state ensembles from a dense network of tight averaged distance restraints derived from exact measurements of nuclear Overhauser enhancements (eNOEs) [Vögeli 2014, Vögeli et al. 2016].
Here, we present the identification of conformational networks harbored by the two human cis/trans isomerases cyclophilin A and Pin1 using the ‘nanorulers’ provided by eNOEs.
We have previously presented an eNOE-based ensemble description of cyclophilin that reveals the presence of a closed and an open state, the latter of which preorganizes the catalytic site for catalysis [Chi et al. 2015]. Based on this finding, we demonstrate here a ligand-selective change of the binding affinity to the active site by tuning of the dynamics of a highly flexible loop [Vögeli et al. 2016]. We show that the binding affinity is increased upon substitution of double glycines to alanines at either of the hinge regions of a loop. The equilibrium distribution is shifted towards more binding-competent conformations.
Comparison of the eNOE-based ensembles of the free and ligand-bound WW domain of Pin1 reveals a conformational network that extends into the interface formed with the enzymatically active PPIase domain. This finding may offer an atomic-picture explanation for the previously discovered communication between the two domains [Peng 2015].
References:
- Vögeli B, Kazemi S, Güntert P, Riek R (2012) Spatial elucidation of motion in proteins by ensemble-based structure calculation using exact NOEs. Nat Struct Mol Biol 19:1053-1057.
- Vögeli B, Olsson S, Güntert P, Riek R (2016) The exact NOE as an alternative in ensemble structure determination. Biophys J 110:113-126.
- Chi C, Vögeli B, Bibow S, Güntert P, Riek R (2015) A structural ensemble for the enzyme cyclophilin reveals an orchestrated mode of action at atomic resolution. Angew Chem Int Ed Engl 54:11657-11661.
- Vögeli B, Bibow S, Chi C (2016) Enzyme selectivity fine-tuned through dynamic control of a loop. Angew Chem Int Ed Engl 55:3096-30100.
- Peng J (2015) Investigating dynamic interdomain allostery in Pin1. Biophys Rev 7:239-249
Shin-ichi Tate
Hiroshima University, JAPAN
Title: Inter-domain communication through intrinsically disordered region (IDR) revealed through the ensemble structure analysis
Time :

Biography:
Abstract:
The functionally relevant inter-domain communication between the domains linked by intrinsically disordered region (IDR) was explored by NMR in combination with small angle X-ray scattering (SAXS). Based on the ensemble structure analyses and the numerical simulations to reproduce the chemical shift changes along with the substrate concentration, we have demonstrated how the domains cooperate to enhance the protein function through the substantially dynamic spatial allocation of the domains.
Pin1, a proline cis/trans isomerase, comprises two domains linked by 10-residue IDR; one is the substrate biding domain to recognize pSer/pThr-Pro motif and the other is the enzyme domain that rotates the Pro peptide bond in the motif. The enzyme domain shows very limited affinity to the substrate, but its binding ability was enhanced by two orders of magnitude in the presence of the substrate binding domain linked by IDR; in which the inter-domain ‘fly-casting’ mechanism plays to keep the substrate bound to Pin1 by tossing and receiving the substrate between the domains, once the substrate in bound to either one of the domains. A new functional aspect of IDR will be addressed.
References:
- Tochio,N., Umehara,K., Uewaki,J., Flechsig,H., Kondo,M., Dewa,T., Sakumar,T., Yamamoto,T., Saitoh,T, Togashi,Y., and Tate,S. (2016): Non-RVD mutations that enhance the dynamics of the TAL repeat array along the superhelical axis improve TALEN genome editing efficacy, Scientific Reports, 6, 37887.
- Wang,J., Tochio,N., Kawasaki,R., Tamari,Y., Xu,N., Uewaki,J., Utsunomiya-Tate, N., and Tate,S. (2015): Allosteric breakage of the hydrogen bond within the dual-histidine motif in the active site of human Pin1 PPIase, Biochemistry, 54, 5242-5253.
- Xu,N. Tochio,N., Wang,J., Tamari,Y., Uewaki,J. Utsunomiya-Tate,N., Igarashi,K., Shiraki,T., Kobayashi,N., and Tate,S. (2014): The C113D mutation in human Pin1 causes allosteric structural changes in the phosphate binding pocket of the PPIase domain through the tug of war in the dualhistidine motif, Biochemistry, 53, 5568-5578.
- Hashimoto,M., Kodera,N., Tsunaka,Y., Oda,M., Tanimoto,M., Ando,T., Morikawa,K., and Tate,S. (2013): Phosphorylation-coupled intramolecular dynamics of unstructured regions in chromatin remodeler FACT, Biophys,J. 104, 2222-2234.
- Mizuno, S., Amida, H., Kobayashi, N., Aizawa, S. and Tate, S. (2011): The NMR structure of FliK, the trigger for the switch of substrate specificity in the flagellar type â…¢ secretion apparatus. J. Mol. Biol., 409, 558-573
Arieh Zaritsky
Ben-Gurion University of the Negev, Israel
Title: Is Nucleoid Complexity hence Cell Diameter Limited by the Eclipse?
Biography:
Arieh Zaritsky of Ben-Gurion University's Faculty of Natural Sciences (http://ariehz.weebly.com/) runs a laboratory investigating parallel fields, pure and applied. During his career at BGU (1973-todate), Dr. Zaritsky has instructed over 50 trainees (graduate students and scientists) and was awarded numerous research grants, allowing him to study both fields of expertise. He visited many higher education Institutions around the world and delivered invited lectures related to both research fields at international meetings. After obtaining a distinguished MSc in Genetics at The Hebrew University of Jerusalem (1967), he graduated at Leicester University (1971) and post-doc'ed at The Copenhagen's Institute of Microbiology (1972). Professor Zaritsky is a recognized expert on bacterial cell physiology and bacteriophage multiplication and published over 130 peer-reviewed articles (http://ariehz.weebly.com/articles.html). Dr. Zaritsky Chaired BGU's Life Sciences department (1989-1991) and is an Editorial Board member of Bioengineered and awardee of 1994 Burroughs-Wellcome/ASM Visiting Professorship.
Abstract:
Cell width W of Escherichia coli is correlated with the mean complexity of its nucleoid, which is expressed as the ratio between the mean times to replicate it and to duplicate the cell aka the number of replication positions n. A set of old, puzzling observations of cell size and dimensions is qualitatively consistent with the view that W is determined by n, and that branching results from breaching a maximum possible value. This maximum nmax is interpreted in terms of a minimal distance possible between successive moving replisomes, so-called eclipse. The data is subject to analytical quantification designed to model the correlations in a way that may (1) shed light on the necessary coupling between the two unique structures in a bacterial cell, nucleoid and sacculus, and (2) lead to decipher the primary signal transduced from DNA to the peptidoglycan biosynthetic pathway. The first approximation is not sufficient to account for the rate at which average cell size rises with time (Po-Yi H and Amir A, personal communication), hence two additional causes are considered to reconcile this discrepancy: loss of division capacity of some DNA-less cells and dependence of the time needed for division on W. A physical signal is invoked, related to transcription/translation of membrane protein genes coupled to membrane-insertion of these proteins termed "transertion", but means to measure the reciprocal stress imposed by transertion strings on both nucleoid and cell envelope are sorely lacking.
Brian Kloss
New York Structural Biology Center, USA
Title: Structural genomics of integral membrane proteins - past successes and future directions

Biography:
Brian Kloss began his research career as a graduate student in the laboratory of Carter Bancroft at the Mount Sinai School of Medicine, studying the transcriptional regulation of the pituitary-specific prolactin and growth hormone genes. He went on to do a postdoc with Michael Young at Rockefeller University, studying the genetic control of circadian rhythms in Drosophila melanogaster. Afterwards, Brian spent almost six years at a biotech startup, helping to develop a cell-based assay for the screening of ligands of GPCRs. For the past ten years, Brian has been a part of the protein production facility of the Center on Membrane Protein Production and Analysis (COMPPÅ), located at the New York Structural Biology Center. There, he has led a small group focused on the identification, cloning and expression screening of integral membrane proteins of prokaryotic origin, mainly for structural studies.
Abstract:
Approximately one-third of all human genes - as well as genes from most other organisms, across all kingdoms of life - encode integral membrane proteins. Nonetheless, the number of integral membrane protein structures solved lags far behind the number of those solved for their soluble counterparts, due primarily to the difficulty of recombinant expression and the instability of membrane proteins once they are detergent-extracted from the lipid bilayer. Over the past 10-20 years, the number of integral membrane protein structures solved, primarily by X-ray crystallography, has increased significantly and structural genomics approaches have played a considerable role in this progress. More recently, advances in cryo-electron microscopy techniques have permitted structures of integral membrane proteins to be determined at resolutions comparable to that of x-ray crystallography, but requiring much smaller quantities of protein. Concurrently, detergents that improve the stability of integral membrane proteins and purification techniques that allow proteins to be extracted and purified in their native lipid environment have also been developed, allowing structural studies of integral membrane proteins to move forward at an exceedingly rapid pace. I will summarize our past integral membrane protein structural biology efforts that employed structural genomics approaches and high-throughput techniques and describe our plans for future structural studies that will continue to make use genomics-based methods, as well as more recently available reagents, techniques and technologies.
References:
- Su et. al. Structural basis for conductance through TRIC cation channels. Nature Communications 19: 15103 (2017).
- Petrou et. al., Structures of aminoarabinose transferase ArnT suggest a molecular basis for lipid A glycosylation. Science 351: 608-612 (2016).
- Ardiccioni et. al., Structure of the polyisoprenyl-phosphate glycosyltransferase GtrB and insights into the mechanism of catalysis. Nature Communications 7: 10175 (2016).
- Beltrán et. al., Control of carotenoid biosynthesis through a heme-based cis-trans isomerase. Nature Chemical Biology 8: 598-605 (2015).
- Guo et. al., Structure and activity of tryptophan-rich TSPO proteins. Science 347: 551-555 (2015).
Steven Hayward
University of East Anglia, UK
Title: Geometrical Principles of Homeric β-Barrels and β-Helices: Applications to Modelling Amyloid Protofilaments

Biography:
Steven Hayward is Reader in Computational Biology at the University of East Anglia. He uses computational methods, including bioinformatics techniques and simulation, to understand protein structure, dynamics and function. He has focused particularly on protein dynamics and developed the popular DynDom method for the analysis of domain movements in proteins (www.cmp.uea.ac.uk/dyndom). Recently he has worked on the fundamental structure of amyloid fibrils and also works on the development of interactive tools for protein visualization and docking using haptics (www.haptimol.com).
Abstract:
Examples of homomeric β-helices and β-barrels have recently emerged. Here we have generalized the theory for the shear number in β -barrels to encompass β-helices and homomeric structures. We introduce the concept of the “β-strip” which comprises neighboring strands, parallel or antiparallel and forms the repeating unit that builds the helix. In this context the shear number is interpreted as the sum of register shifts between neighboring β-strips. This more general approach has allowed us to derive relationships between the helical width, helical pitch, angle between strand direction and helical axis, mass per length, register shift, and number of strands. The validity and unifying power of the method is demonstrated with known structures including the T4 phage spike, cylindrin, and the HET-s(218-289) prion. The relationships have allowed us to predict register shift and number of strands in transthyretin and Alzheimer β(40) amyloid protofilaments from reported dimensions measured by X-ray fiber diffraction which we have used to construct models that comprise a single strip of in-register β-strands folded into a “β-strip helix”. The results suggest that both stabilization of an individual β-strip helix as a protofilament subunit and growth of the protofilament by the joining of subunits end-to-end, would involve the association of the same pair of sequence segments at the same register shift.

T4 phage spike
References:
- S Hayward, DP Leader, F Alâ€Shubailly, EJ Milnerâ€White (2014) Rings and ribbons in protein structures: Characterization using helical parameters and Ramachandran plots for repeating dipeptides. Proteins 82 (2): 230-239.
- S.Hayward, E.J.Milner-White (2011) Simulation of the β- to α-sheet transition results in a twisted sheet for antiparallel and an α-nanotube for parallel strands: Implications for amyloid formation. Proteins 79(11): 3193-3207.
- D Taylor, G Cawley, S Hayward (2014) Quantitative method for the assignment of hinge and shear mechanism in protein domain movements. Bioinformatics 30(22): 3189-3196.
- S. Hayward and A. Kitao (2015) Monte Carlo Sampling with Linear Inverse-Kinematics for Simulation of Protein Flexible Regions. Journal of Chemical Theory and Computation 11 (8): 3895-3905.
- Georgios Iakovou, Steven Hayward, Stephen Laycock (2017) A virtual environment for studying the docking interactions of rigid biomolecules with haptics. Journal of Chemical Information and Modeling 57 (5): 1142–1152.
Enrica Bordignon
Ruhr-Universität Bochum, Germany
Title: Exploring conformational equilibria of a heterodimeric ABC transporter by electron paramagnetic resonance
Time :
Biography:
Enrica Bordignon is an associate professor at the Ruhr University of Bochum, where she leads an EPR laboratory dedicated to the study of membrane proteins. Approximately 50 papers in protein research by EPR methods. Research interests: understanding the mechanism of action of proteins by EPR.
Abstract:
ABC exporters pump substrates across the membrane by coupling ATP- driven movements of nucleotide binding domains (NBDs) to the transmembrane domains (TMDs), which switch between inward- and outward-facing (IF and OF) orientations. Understanding their cycle has potential for medical applications because they are involved in multidrug resistance of cancer cells.
Site-directed spin labeling electron paramagnetic resonance (EPR), and in particular the dipolar spectroscopy technique called DEER (or PELDOR) was used to investigate the conformational transition of the ABC heterodimeric exporter TM287/288 from the hyperthermophile T. maritima. The analysis revealed that with nucleotides the transporter exists in an equilibrium between the IF and OF states. ATP binding without hydrolysis was sufficient to partially populate the OF state, and an almost complete conformational shift was observed when nucleotides were trapped in a pre- or post-hydrolytic state. At physiological temperature and without nucleotides, the NBDs disengage asymmetrically while the conformation of the TMDs remains unchanged. Nucleotide binding at the degenerate ATP site prevents complete NBD separation, a molecular feature differentiating heterodimeric ABC exporters from their homodimeric counterparts. Our data suggest hydrolysis-independent partial closure of the NBD dimer, which is further stabilized as the consensus site nucleotide is committed to hydrolysis. A unified mechanism is established, which reconciles the available information for heterodimeric ABC exporters1.
References:
- Timachi MH, Hutter CAJ Hohl M, Assafa T, Böhm S, Mittal A, Seeger MA, Bordignon E (2017) Exploring conformational equilibria of a heterodimeric ABC transporter. eLife 6:e20236..
- Celia H, Noinaj N, Zakharov SD, Bordignon E, Botos I, Santamaria M, Barnard TJ, Cramer WA, Lloubes R, Buchanan SK (2016) Structural insight into the role of the Ton complex in energy transduction. Nature 538 (7623):60-65.
- de Almeida Ribeiro E, Pinotsis N, Ghisleni A, Salmazo A, Konarev PV, Kostan J, Sjöblom B, Schreiner C, Polyansky AA, Gkougkoulia EA, Holt MR, Aachmann FL, Žagrović B, Bordignon E, Pirker KF, Svergun DI, Gautel M, Djinović-Carugo K (2014) The structure and regulation of human muscle α-actinin. Cell 159 (6):1447-1460
- Bleicken S, Jeschke G, Stegmueller C, Salvador-Gallego R, García- Sáez AJ, Bordignon E (2014) Structural model of active Bax at the membrane. Molecular Cell 56 (4):496-505.
- Hohl M, Hürlimann LM, Böhm S, Schöppe J, Grütter MG, Bordignon E, Seeger MA (2014) Structural basis for allosteric cross-talk between the asymmetric nucleotide binding sites of a heterodimeric ABC exporter. PNAS 111 (30):11025-11030.
J. Chakrabarti
S. N. Bose National Centre for Basic Sciences, India
Title: Microscopic calculation of conformational thermodynamics in bio-macromolecular complexes
Biography:
Jaydeb Chakrabarti is a theoretical physicist trained in condensed matter physics. He looks into Statics and Dynamics of soft matter systems including bio-macromolecules. The theoretical methods used in these studies are: (1) Computer simulations based on Molecular Dynamics, Monte Carlo and Brownian Dynamics; (2) Mean field calculations based on classical density functional theories. The goal of his researches is to relate macroscopic properties to microscopic motions.
Abstract:
Statement of the Problem: The microscopic basis of connection between protein conformation and function is a fundamental challenge. Recent experiments show the importance of conformational changes in providing stability to protein complexes. The changes in conformational state have both enthalpy and entropic components. It has been possible to quantify the entropy changes due conformational changes from Nuclear magnetic Resonance data. Here we show microscopic calculation of both the enthalpy and entropy contribution while protein conformations change from the equilibrium distribution of the dihedral angles of proteins. We have shown that the free-energetically destabilized and entropically disordered residues in a given conformation compared to a reference conformation act as binding residue in the giv en conformation. Here we show that this principle can: (1) ascertain ligand binding of a protein in different conformations; (2) supplement the structure of missing fragments in a protein; and (3) serve as a guideline for allosteric changes. All these applications point to tune protein in-silico which would help to design functional materials with protein as building blocks.
References:
- Conformational thermodynamics guided structural reconstruction of biomolcular fragments, Samapan Sikdar, J. Chakrabarti and Mahua Ghosh, Molecular Biosystems, 12, 444-453 (2016).
- A microscopic insight from conformational thermodynamics to functional ligand binding in proteins, Samapan Sikdar, J. Chakrabarti and Mahua Ghosh, Molecular Biosystems, 10, 3280-3289 (2014).
- Quantum chemical studies on the role of residues in calcium binding to Calmodulin, Samapan Sikdar, Mahua Ghosh, Molly De Raychaudhury and J. Chakrabarti, Chem Phys Lett, 605-606, 103-107(2014).
- Conformational contribution to thermodynamics of binding in protein-peptide complexes through microscopic simulation Amit Das, J. Chakrabarti and Mahua Ghosh, Biophysical Journal, 104, 1274 (2013).
- High-Affinity Quasi-Specific Sites in the Genome: How the DNA-Binding Proteins Cope with them J. Chakrabarti, Navin Chandra, Paromita Raha, and Siddhartha Roy, Biophysical Journal, 101, 1123 (2011).
- Track 10: Signalling Biology
Session Introduction
Irena Levitan
University of Illinois at Urbana–Champaign, USA
Title: Structural insights into cholesterol regulation of inwardly-rectifying K+ channels

Biography:
Irena Levitan has done her PhD and is a Professor of Medicine and Adjunct Professor of Bioengineering at the University of Illinois at Chicago. Her current research focuses on cholesterol regulation of ion channels and cellular biomechanics. Her group has provided the first comprehensive structural insights into cholesterol regulation of K+ channels and the cross-talk between cholesterol and other regulators of these channels. She was named a Guyton Distinguished Lecturer by the Association Chairs of Departments of Physiology for her quantitative and biophysical work on cholesterol modulation of ion channels and how this can affect integrated organ function. She is an author of more than 70 publications and a leading Editor of Cholesterol Regulation of Ion Channels and Receptors (Wiley, 2012) and Vascular Ion Channels in Physiology and Disease (Springer, 2016).
Abstract:
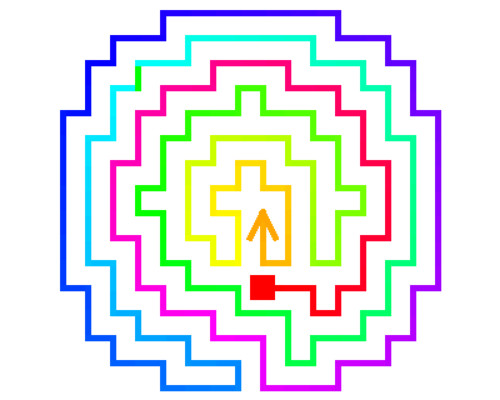
Cholesterol is known to play a major role in regulating the function of multiple membrane proteins including a growing number of ion channels. Our studies focus on inwardly-rectifying K+ (Kir) channels that are ubiquitously expressed in mammalian cells and are known to play major role in membrane excitability and shear stress sensation. In this study, we have shown that Kir channels are suppressed by loading the cells with cholesterol and enhanced by cholesterol depletion. A series of studies revealed that cholesterol interacts with the channels directly by stabilizing them in a long-lived closed “silent” state and that multiple structural features of the channels are essential for conferring their cholesterol sensitivity. Using a combined computational-experimental approach, we show that cholesterol may bind to two nonannular regions that form hydrophobic pockets between the transmembrane helices of the adjacent subunits of the channel. The location of the binding regions suggests that, cholesterol modulates channel function by affecting the hinging motion at the center of the porelining transmembrane helix that underlies channel gating. In addition, we identified a series of residues in the C and N-terminus of the channel. These are critical for conferring cholesterol sensitivity to the channels, but are not part of the binding sites. These residues form a distinct cytosolic structure, a cholesterol sensitivity belt which surrounds the cytosolic pore of the channel in proximity to the transmembrane (TM) domain, and includes residues whose mutation results in abrogation of the channel’s cholesterol sensitivity. Further analysis identified a reversal residue chain comprised of residues that link one of the cholesterol sensitivity belt residues with a distant cytosolic residue that constitute a two-way molecular switch of the channel sensitivity to cholesterol. Further studies are needed to elucidate the connection between cholesterol binding and channel.
References:
- Rosenhouse-Dantsker A, Noskov S, Durdagi S, Logothetis DE, Levitan I. Identification of novel cholesterol-binding regions in Kir2 channels. JBC, 288(43):31154-64, 2013
- Han H., Rosenhouse-Dantsker A. Gnanasambandam R. Epshtein Y. Chen Z., Sachs F, Minshall RD and I Levitan. Silencing of Kir2 channels by caveolin-1: cross-talk with cholesterol. Journal of Physiology, 592:4025-38, 2014
- Rosenhouse-Dantsker A. Epshtein Y. and I. Levitan. Interplay between lipid modulators of Kir2 channels: cholesterol and PIP2. Computational and Structural Biotech Journal, 11(19):131-7, 2014
- Bukiya AN, Osborn CV, Kuntamallappanavar G, Toth PT, Baki L, Kowalsky GB, Oh MO, Dopico AM, Levitan I. and A. Rosenhouse-Dantsker. Cholesterol increases the open probability of cardiac KACh currents, BBA Membranes, 848:2406-13, 2015
- Ahn SJ, Fancher IS, Bian JT, Zhang CX, Schwab S, Gaffin R, Phillips SA, and I Levitan. Inwardly-rectifying K+ channels are major contributors to flow-induced vasodilation in resistance arteries. J of Physiology. In Press.
Joachim Krebs
Max Planck Institute for Biophysical Chemistry, Germany
Title: Calcium, Calmodulin and the Plasma Membrane Calcium Pump

Biography:
Prof. RJP Williams at Oxford, UK. In 1977 he accepted a staff position at the Institute of Biochemistry at the Swiss Federal Institute of Technology (ETH) in Zürich, Switzerland. He was lecturing different courses in biochemistry and biophysics, and was leading a Lab working on the structure-function relationship of calcium-binding and calcium-transporting proteins. After retirement from the ETH he continued his research as a consultant of the Lab of Prof. Christian Griesinger at the MPI in Göttingen, Germany. He has authored, co-authored and edited numerous articles in international journals. Recently he edited a book on “Calcium: A matter of life or death” published in 2007. He is at the Editorial Board of BBA Molecular Cell Research and Archives of Biochemistry and Biophysics.
Abstract:
Calcium is the third most abundant metal in nature and a versatile carrier of many signals within and outside the cell. Due to its peculiar coordination chemistry calcium is highly flexible as a ligand which enables it to regulate many important aspects of cellular activity. Calcium can fulfill its many different functions insite and out of the cell due to an integrated network of calcium channels, exchangers and pumps. In this presentation I will give an overview on our studies of calcium binding proteins, their interaction with protein targets resulting in specific modulations of protein-protein interactions. This will be demonstrated by the interaction of the calcium binding protein calmodulin with one of its targets, the plasma membrane calcium pump, an important regulator of calcium homeostasis of the cell.
References:
- Krebs, J. (2015) The plethora of PMCA isoforms: Alternative splicing and differential expression. Biochim. Biophys. Acta 1853: 2018-2024
- Seidel, K. et al. (2008) Structural characterization of Ca(2+)-ATPase-bound phospholamban in lipid bilayers by solid-state nuclear magnetic resonance (NMR) spectroscopy. Biochemistry 47: 4369-4376.
- Elshorst, B., Krebs, J. et al. (1999) NMR solution structure of a complex of calmodulin with a binding peptide of the Ca2+-pump. Biochemistry 38: 12320-12332.
- Carafoli, E., Krebs, J. (2016) Why Calcium? How Calcium became the best Communicator. J. Biol. Chem. 291: 20849-20857.
5.Toyoshima, C. (2009) How Ca2+-ATPase pumps ions across the sarcoplasmic reticulum membrane. Biochim. Biophys. Acta 1793: 941-946.
Carol A. Heckman
Bowling Green State University, USA
Title: Structural aspects of cell signaling
Time :

Biography:
Carol Heckman is an expert on preneoplasia and image analysis and has developed optical and computational methods suitable for cell feature analysis. Applying these methods, she showed how to deconstruct the cell phenotype and find the features related to oncogenic transformation. About half of the 20 features are useful in distinguishing the phenotypes of normal and cancerous epithelial cells. She has published numerous papers on these topics. The cell features form the basis of an assay to flag chemicals of interest for drug development and identify diseases that can be targeted productively by existing drugs.
Abstract:
Statement of the problem: Endpoints such as adhesion and motility have been used to infer the function of a protein in cells. These endpoints are unsatisfactory, because a protein can be recruited to different substructures and promote different outcomes in such structures. By defining meaningful endpoints, it is possible to identify a protein’s contribution to several different patterns of cell organization and thereby address major problems in biology.
Methodology and theoretical orientation: We developed an unbiased method of classifying and quantifying features of fixed, adherent epithelial cells. Primary data, consisting of 102 measures of contour geometry, curvature, relationship to derived model figures, etc., were used to calculate 20 latent factors representing cell features. Factors detect structure by recognizing the relationships between variables. Cells from experiments are classified according to each factor by summing the factor loadings. Filopodia (factor 4) accounted for a larger proportion of cancer-related variance than any other feature. Filopodia are the sensory appendages that are relied on when cells distinguish their more and less adhesive sides. The protrusion defined as factor 7 represented neurites. Even when small, neurites differed from lamellipodia (factor 5). Several factors contribute to ruffling.
Findings: Filopodia are down-regulated by three isoforms of protein kinase C (PKC). The effect of PKC ε, a known oncogene, on filopodia is only observed after tumor promoter treatment. The effect is in part due to a PKC ε-mediated increase in ruffling. In cells not treated with tumor promoter, filopodia are down-regulated by isoforms a and h. PKC a has contrary effects in promoter-treated cells, where it conserves filopodia by suppressing ruffling activity. Activated PKC a may promote filopodia. These activities are consistent with the concept that filopodia are implicated in cell homeostasis. By regulating the prevalence of filopodia, PKC can regulate the way cells react to their surroundings.
References:
- Heckman CA, Pandey P, Cayer ML, Biswas T, Zhang ZY, Boudreau NS (2017) The tumor promoter-activated protein kinase Cs are a system for regulating filopodia. Cytoskeleton (Hoboken) May 8. doi: 10.1002/cm.21373. [Epub ahead of print]
- Mukhopadhyay C, Triplett A, Bargar T, Heckman C, Wagner K-, M Naramura M (2016) Casitas B-cell lymphoma (Cbl) proteins protect mammary epithelial cells from proteotoxicity of active c-Src accumulation. PNAS USA 113: E8228-E8237
- Amarachintha SP, Ryan KJ, Cayer M, Boudreau NS, Heckman CA (2014) Effect of Cdc42 domains on filopodia sensing, cell orientation, and haptotaxis. Cellular Signalling S0898-6568(14)00379-9. doi: 10.1016/j.cellsig.2014.11.025.
- Heckman CA, Plummer HK III (2013) Filopodia as sensors. Cellular Signaling 25: 2298-2311. doi: 10.1016/j.cellsig.2013.07.006.
- Heckman CA, Varghese M, Cayer ML, Boudreau NS (2012) Origin of ruffles: Linkage to other protrusions, filopodia and lamellae. Cellular Signaling 24: 189-198.
Vesa P Hytönen
University of Tampere, Finland
Title: Mechanical Stability of Talin Rod Controls Traction Force Generation and Cell Migration

Biography:
Vesa P Hytönen is a Head of the Protein Dynamics research group in BioMediTech at the University of Tampere, Tampere, Finland. After graduating as a PhD from the University of Jyväskylä, Jyväskylä, Finland in 2005, he has conducted Post-doctoral training at ETH Zurich, Zürich, Switzerland from 2005-2007. He then continued as a Post-doctoral researcher at the University of Tampere and established independent research group in 2010. He is currently working as Associate Professor at the University of Tampere. His research interests are mechanobiology, protein engineering and vaccine research and authored more than 100 scientific articles.
Abstract:
Talin is a central adhesion protein linking β-integrin cytosolic domains to actin fibers. It participates in the transmission of mechanical signals between extracellular matrix and cell cytoskeleton. Talin rod domain consists of a series of mechanically vulnerable α-helical subdomains containing binding sites for other adhesion proteins such as vinculin, actin and RIAM. Force induced unfolding of these rod subdomains has been proposed to act as a cellular mechanosensor, but so far evidence linking their mechanical stability and cellular response has been lacking. We show that stepwise mechanical destabilization of talin rod subdomain increases talin and vinculin accumulation into cell-matrix adhesions and decreases cell migration rate. In addition, mechanical destabilization of talin subdomain was found to decrease cellular traction force generation and to promote the formation of adhesions on fibronectin over vitronectin. Experiments with truncated talin forms confirmed the mechanosensory role of the talin subdomain and excluded the possibility that the observed effects are caused solely by the release of talin autoinhibition. We demonstrate that by modulating the mechanical stability of an individual talin rod sub-domain, it is possible to affect traction force generation, ECM sensing and consequently highly coordinated processes such as cell migration. Our results suggest that talin acts as a mechanosensor and is responsible for controlling the cellular processes dependent on mechanical signals and cellular mechanosensing.
References:
- von Essen M, Rahikainen R, Oksala N, Raitoharju E, Seppälä I, Mennander A, Sioris T, Kholová I, Klopp N, Illig T, Karhunen PJ, Kähönen M, Lehtimäki T, Hytönen VP (2016) Talin and vinculin are downregulated in atherosclerotic plaque; Tampere Vascular Study. Atherosclerosis. 255:43-53.
- Qi L, Jafari N, Li X, Chen Z, Li L, Hytönen VP, Goult BT, Zhan CG, Huang C (2016) Talin2-mediated traction force drives matrix degradation and cell invasion. J Cell Sci. 129:3661-3674.
- Haining AW, von Essen M, Attwood SJ, Hytönen VP, Del Río Hernández A (2016) All Subdomains of the Talin Rod Are Mechanically Vulnerable and May Contribute To Cellular Mechanosensing. ACS Nano 10:6648-58.
- Hytönen VP, Wehrle-Haller B (2016) Mechanosensing in cell-matrix adhesions - Converting tension into chemical signals. Exp Cell Res. 343:35-41.
- Hytönen VP, Vogel V (2008) How force might activate talin's vinculin binding sites: SMD reveals a structural mechanism. PLoS Comput Biol. 4:e24.
Biography:
Francis Millett received his B.S in Chemistry from the University of Wisconsin in 1965, his Ph.D. in Chemical Physics from Columbia University in 1970, and was an NIH Postdoctoral Fellow at California Institute of Technology from 1970-1972. He joined the faculty of the University of Arkansas in 1972, and is now a Distinguished Professor. He developed, together with Bill Durham, the ruthenium photoreduction method which made it possible to measure the kinetics of key steps in electron transfer during mitochondrial oxidative phosphorylation. He has directed collaborative, multidisciplinary research which combines rapid kinetics methods, site-directed mutagenesis, X-ray crystallography, and NMR to investigate protein structure-function relationships.
Abstract:
The electron transfer reactions within wild-type Rhodobacter sphaeroides cytochrome bc1 (cyt bc1) were studied using a ruthenium dimer to rapidly photooxidize cyt c1. It was found that when cyt bH was initially reduced before the reaction, photooxidation of cyt c1 led to bifurcated reduction of both the iron-sulfur protein and cyt bL by QH2 in the Qo site, followed by re-oxidation of two equivalents of cyt bL and cyt bH. It was proposed that the newly formed ubiquinone diffused through the hydrophobic cavity linking the Qo site of the reactive monomer A to the Qi site of the other monomer B, leading to oxidation of cyt bH in monomer B followed by oxidation of cyt bL in monomer A by cross-monomer electron transfer. Addition of one equivalent of the Qi site inhibitor antimycin to the cyt bc1 dimer had very little effect on any of the electron transfer reactions, while addition of a second equivalent completely inhibited re-oxidation of cyt bL and cyt bH. It was also found that addition of one equivalent of the Qo site inhibitor stigmatellin to the cyt bc1 dimer completely inhibited all electron transfer reactions in both monomers of the dimer. These experiments are consistent with a half-of-the-sites mechanism in which only one monomer of the dimer is active at a time, implying monomer-monomer interactions. The rapid electron transfer reaction from the ISP to cyt c1 was found to be greatly decreased by viscosity, indicating a multi-step diffusional mechanism as the iron-sulfur protein rotates from the b state to the c1 state.
References:
- Janzon, Julia; Yuan, Quan; Malatesta, Francesco; Hellwig, Petra; Ludwig, Bernd; Durham, Bill; Millett, Francis. “Probing the Paracoccus denitrificans cytochrome c1-Cytochrome c552 interaction by mutagenesis and fast kinetics,” Biochemistry (2008) 47(49), 12974-12984.
- Mills, D.Z.; Xu, Shujuan; Geren, L.; Hiser, C.; Qin, L.; Sharpe, M.A.; McCracken, J.; Durham, B.; Millett, F.; Ferguson-Miller, S. Proton-Dependent Electron Transfer from CuA to Heme a and Altered EPR Spectra in Mutants Close to Heme a of Cytochrome Oxidase. Biochemistry (2008), 47(44), 11499-11509.
- Castellani, M.; Havens, J.; Kleinschroth, T.; Millett, F.; Durham, B.; Malatesta, F.; Ludwig, B. “The acidic domain of cytochrome c(1) in Paracoccus denitrificans, analogous to the acidic subunits in eukaryotic bc(1) conmplexes, is not involved in the electron transfer reaction to its native substrate cytochrome c(552). Biochim Biophys Acta (2011), 1807:1383-1389
- Havens, J.; Castellani, M.; Kleinschroth, T.; Ludwig, B.; Durham, B.; Millett, F. Photoinitiated electron transfer within the paracoccus denitrificans cytochrome bc(1) complex: Mobility of the iron-sulfur protein is modulated by the occupant of the Q(o) site. Biochemistry (2011) Dec 6;50(48):10462-10472.
- Durham B, Millett F. “Design of photoactive ruthenium complexes to study electron transfer and proton pumping in cytochrome oxidase.” Biochim Biophys Acta. (2012) Apr;1817(4):567-74.
Leonas Valkunas
Vilnius university, Lithuania
Title: Mapping energy transfer channels in fucoxanthin–chlorophyll protein complex

Biography:
Leonas Valkunas is the author of more than 350 international publications, head of the department of Theoretical Physics at Vilnius university and head of the Department of Molecular Compounds Physics at the Center for Physical Sciences and Technology in Vilnius. He is involved in studies of primary processes of photosynthesis such as excitation dynamics and photoinduced charge separation in various photosynthetic systems based on the spectroscopic data and using various theoretical modelling approaches.
Abstract:
Fucoxanthin–chlorophyll protein (FCP) is the key molecular complex performing the light-harvesting function in diatoms, which, being a major group of algae, are responsible for up to one quarter of the total primary production on Earth. These photosynthetic organisms contain an unusually large amount of the carotenoid fucoxanthin, which absorbs the light in the blue–green spectral region and transfers the captured excitation energy to the FCP bound chlorophylls. Due to the large number of fucoxanthins, the excitation energy transfer cascades in these complexes are particularly tangled. Energy transfer processes and coherent phenomena in the fucoxanthin–chlorophyll protein complex, which is responsible for the light harvesting function in marine algae diatoms, were investigated at 77 K by using two-dimensional electronic spectroscopy. Experiments performed on femtosecond and picosecond timescales led to separation of spectral dynamics, witnessing evolutions of coherence and population states of the system in the spectral region of Qy transitions of chlorophylls a and c. Analysis of the coherence dynamics allowed us to identify chlorophyll (Chl) a and fucoxanthin intramolecular vibrations dominating over the first few picoseconds. Closer inspection of the spectral region of the Qy transition of Chl c revealed previously not identified, mutually non-interacting chlorophyll c states participating in femtosecond or picosecond energy transfer to the Chl a molecules. Consideration of separated coherent and incoherent dynamics allowed us to hypothesize the vibrations-assisted coherent energy transfer between Chl c and Chl a and the overall spatial arrangement of chlorophyll molecules.
References:
- Butkus V, Gelzinis A, Augulis R, Gall, A, Büchel C, Robert B, Zigmantas D, Valkunas L, Abramavicius D (2015) Coherence and population dynamics of chlorophyll excitation in FCP complex: two-dimensional spectroscopy study. J. Chem. Phys. 142: 212414.
- Gelzinis A, Butkus V, Songaila E, Augulis R, Gall A, Büchel C, Robert B, Abramavicius D, Zigmantas D, Valkunas L (2015) Mapping energy transfer channels in fucoxanthin-chlorophyll protein complex. Biochim. Biophys. Acta 1847: 241-247.
- Farooq S, Chmeliov J, Trinkunas G, Valkunas L, van Amerongen H. (2016) Is there excitation energy transfer between different layers of stacked photosystem II containing thylakoid membranes? J. Phys. Chem. Lett.7: 1406-1410.
- Chmeliov J, Gelzinis A, Songaila E, Augulis R, Duffy CDP, Ruban AV, Valkunas L. (2016) The nature of self-regulation in photosynthetic light-harvesting antenna. Nature Plants 2: 16045.
- Abramavicius D, Valkunas L. (2016) Role of coherent vibrations in energy transfer and conversion in photosynthetic pigment-protein complexes. Photosynth. Res. 127: 33-47.

Biography:
Jianmin Cui is a professor on the Spencer T. Olin Endowment at Washington University in St. Louis, in the Department of Biomedical Engineering. He received Ph.D. in Physiology and Biophysics from State University of New York at Stony Brook and a post-doctoral training at Stanford University. He was an assistant professor of Biomedical Engineering at Case Western Reserve University before moving to St. Louis. His research interests include BK-type calcium-activated potassium channels and IKs channels.
Abstract:
Voltage-gated ion channels generate dynamic ionic currents that are vital to the physiological functions of many tissues. These proteins contain separate voltage-sensing domains, which detect changes in transmembrane voltage, and pore domains, which conduct ions. Coupling of voltage sensing and pore opening is critical to the channel function and has been modeled as a protein–protein interaction between the two domains. However, our data show that coupling in Kv7.1 channels requires the lipid phosphatidylinositol 4,5-bisphosphate (PIP2). We found that voltage-sensing domain activation failed to open the pore in the absence of PIP2. This result is due to loss of coupling because PIP2 was also required for pore opening to affect voltage-sensing domain activation. We identified a critical site for PIP2-dependent coupling at the interface between the voltage-sensing domain and the pore domain. This site is actually a conserved lipid-binding site among different K+ channels, suggesting that lipids play an important role in coupling in many ion channels. To further investigate the mechanism of PIP2 mediated VSD-pore coupling, we identified a compound that mimics PIP2 structure and function as a molecular probe. This compound was identified using an in silico screening approach based on molecular docking of a library of compounds to the PIP2 binding site in a homology model of the Kv7.1 channel. Our results show that this compound can substitute PIP2 in activating the Kv7.1 channel.
References:
- Cui, J. (2016) Voltage dependent gating: novel insights from KCNQ1 channels. Biophys J 110:14-25.
- Kasimova M, Zaydman M, Cui J, Tarek M (2015) PIP2-dependent coupling is prominent in Kv7.1 due to weakened S4-S5/S6 interactions. Scientific Reports 5:7474.
- Zaydman, M.A. and Cui, J. (2014) PIP2 activation of KCNQ channels. Front Physiol. 5:195. doi: 10.3389/fphys.2014.00195.
- Zaydman M, Kasimova M, McFarland K, Liang H, Beller Z, Shi J, Hou P, Kinser H, Zhang G, Tarek M, and Cui J (2014) Domain-domain interactions determine the gating, permeation, pharmacology, and subunit modulation of the IKs ion channel. eLife 2014;3:e03606.
- Zaydman MA, Silva JR, Delaloye K, Li Y, Liang H, Larsson HP, Shi J, and Cui J (2013) Kv7.1 ion channels require PIP2 to couple voltage sensing to pore opening. Proc. Natl. Acad. Sci., U.S.A. 110:13180-13185.
Igor L. Barsukov
University of Liverpool, UK
Title: LD Motif Interacting networks in cell-matrix adhesion.
Time :

Biography:
Igor Barsukov has expertise in structural biology, primarily using NMR spectroscopy and X-ray crystallography. The main focus of his research has been on the structure and function of integrin-mediated cell-matrix adhesions, where he directed full structural analysis of the key adhesion proteins talin, leading to the currently widely used model of stretch-dependent talin activation. He is currently extending the model of talin functionality to include competitive, talin based, interaction networks. Recently he identified interactions between neuronal scaffold proteins Shank3 and Ras-family GTPase that regulate integrin activity and have implications for control of synaptic plasticity.
Abstract:
Statement of the Problem: Cell-matrix adhesion requires assembly of large multi-protein complexes linked to the cyto-domains on the integrin adhesion receptors. These complexes dynamically change in response to the adhesion forces and environmental signals. Mechano-sensitive adaptor protein talin couples the force and adhesion signaling by acting as a hub for numerous, often competitive, interactions. The molecular mechanism that controls the co-ordination of the interactions in time and space is currently not understood. The aim of this study is to define the interactions between talin and Rho-GAP Deleted in Liver Cancer 1 (DLC1) that regulates adhesion forces. Methodology: Structure of the talin/DLC1 complex was solved by X-ray crystallography and the interactions between the proteins analysed by NMR spectroscopy. Fluorescent imaging was used to define the interactions within the adhesion complexes and cancer cell lines were employed to characterise the effect of the interaction on the biological activity. Findings: We defined the atomic details of the talin/DLC1 interactions and used this information to identify signalling protein paxillin as a talin ligand. Based on the structural information we designed a range of talin mutants that modulate the interactions and demonstrated that the mutations reduce DLC1 signalling in adhesion. Conclusion & Significance: Talin recognises LD motifs in DLC1 and paxillin through a set of well-defined charge interactions. These interactions are similar to other LD motif interactions previously identified in signalling pathways. These interactions are also similar to the interactions between talin, RIAM and vinculin that were previously not assigned to the LD motif family. Together, the relatively weak LD motif interactions within the adhesion complex create a protein network that could dynamically respond to the adhesion signals.
References:
- Zacharchenko, T., et al., LD Motif Recognition by Talin: Structure of the Talin-DLC1 Complex. Structure, 2016.
- Atherton, P., et al., Vinculin controls talin engagement with the actomyosin machinery. Nat Commun, 2015. 6: p. 10038.
- Goult, B.T., et al., RIAM and Vinculin Binding to Talin Are Mutually Exclusive and Regulate Adhesion Assembly and Turnover. J Biol Chem, 2013. 288(12): p. 8238-8249.
- Goult, B.T., et al., Structural studies on full-length talin1 reveal a compact auto-inhibited dimer: Implications for talin activation. J Struct Biol, 2013. 184(1): p. 21-32.
- Elliott, P.R., et al., The Structure of the Talin Head Reveals a Novel Extended Conformation of the FERM Domain. Structure, 2010. 18(10): p. 1289-1299.
Arfaxad Reyes Alcaraz
Korea University College of Medicine, South Korea
Title: Structural Conformational changes report biased agonism: The case of Galanin receptors

Biography:
Arfaxad Reyes has his expertise in structure and stability of G-protein coupled receptors and passion for improving and creating new drug discovery platforms that greatly contribute in the development of more selective drugs with minor side effects. His studies about biased agonism in Galanin receptors help to understand the relationship between conformational structure of the receptor and its corresponding physiological effect induced by a specific ligand. Recently he and co-workers were able to develop a highly selective agonist for Galanin receptor 2 with anxiolytic effect in vivo (Arfaxad Reyes-Alcaraz et al Sci. Rep 2016) which was the base to discover how different ligand structures induce different conformations on the structure of Galanin receptors. This work greatly contribute to understand the relationship between structure and function of Galanin receptors.
Abstract:
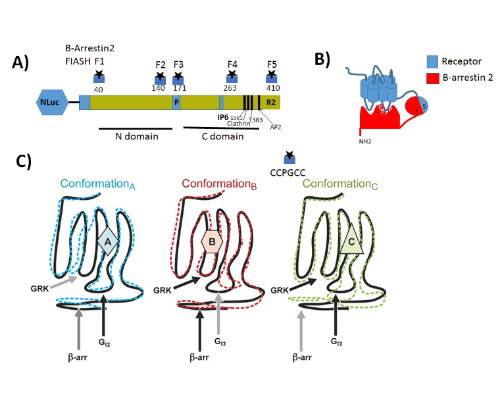
Statement of the Problem: G protein coupled receptors (GPCRs) also known as seven-transmembrane receptors and are the largest family of cell-surface receptors that communicate extracellular stimuli to the cell interior (1). It is now accepted that chemically distinct ligands bind to the same GPCR and can stabilize the receptor in multiple active conformations, which results in differential activation of cell signaling pathways and, eventually, in different physiologic outcomes a phenomenon known as biased agonism (2). Biased agonism can be exploited to design drugs that selectively activate signaling pathways, leading to the desired physiologic effects while on target side effects elicited by activation of other signaling pathways via the same receptor subtype (3). Methodology & Theoretical Orientation: The aim of this study was to stablish a relationship between conformational changes in Galanin receptors and their signaling properties in living cells, for that purpose we develop a structural complementation assay based on NanoBit technology and a series of conformational fluorescein arsenical hairpin (FIASH) bioluminescence resonance energy transfer (BRET) biosensors to monitor structural changes β-arrestin 2 induced by the binding with each Galanin receptor. Findings: Here we show that Galanin receptors impose different conformational signatures in β-arrestin, moreover structurally different ligands activating the same receptor imposed different conformations in β-arrestin 2 producing biased signaling. Conclusion & Significance: Our data provide definite evidence that a receptor activated by structurally different ligands can adopt multiple active conformations. Moreover, this finding also demonstrates that functionally specific structural Galanin receptor conformations can indeed be translated to downstream effectors producing a different physiological response.
References:
- Reyes-Alcaraz A., Lee, Y. N., Son, G. H., Kim, N. H., Kim, D. K., and Yun, S., Kim D. H., Hwang, J. I. and Seong, J. Y. Development of Spexin-based Human Galanin Receptor Type IISpecific Agonists with Increased Stability in Serum and Anxiolytic Effect in Mice. Scientific Reports, 6:21453 (2016).
- Reyes-Alcaraz A., Matinez-Archundia M., Ramon E. and Garriga P. Salt Effects on the Conformational Stability of the Visual G-Protein-Coupled Receptor Rhodopsin. Biophysical Journal, 101, 2798-2806 (2011).
- Moon M. J., Lee Y.-N., Park S., Reyes-Alcaraz A., Hwang J. –I., Millar R. P., Choe H. and Seong J. Y. Ligand Binding Pocket Formed by Evolutionarily Conserved Residues in the Glucagon-like Peptide-1 (GLP-1) Receptor Core Domain. The Journal of Biological Chemistry, 290, 5696-5706, (2015).
- Yun, S., Furlong M., Sim M., Cho M., Park, S., Cho E. B., Reyes-Alcaraz, A., Hwang, J. -I., Kim, J., and Seong J. Y. Prevertebrate Local Gene Duplication Facilitated Expansion of the Neuropeptide GPCR Superfamily. Molecular Biology and Evolution 1-15, (2015).
- Reyes-Alcaraz A., Tzanov T. and Garriga P. Stabilization of Membrane Proteins: the Case of G-Protein-Coupled Receptors. Engineering in Life Sciences, 8, 207–217 (2008).
Alessandra Astegno
University of Verona, Italy
Title: A biophysical and structural approach to investigate calcium sensor properties of plant calmodulin-like proteins
Biography:
Alessandra Astegno is interested in different aspects of protein chemistry, including folding, evolution and structure-function relationship of proteins and macromolecular assemblies. She obtained a PhD in Applied Biotechnologies from University of Verona in 2010. She is currently assistant professor in Biochemistry at the Department of Biotechnology of the University of Verona. She has a solid background in recombinant protein expression and purification, functional and structural characterization of metallo-proteins as well as PLP-dependent enzymes. Recently, her work focused on the study of calcium signaling in higher plants through biophysical, biochemical and structural characterization of calcium sensor proteins, such as calmodulin and calmodulin like proteins of Arabidopsis thaliana.
Abstract:
Calcium is an essential second messenger in plants that regulates various signaling pathways through stimulus-specific Ca2+ signatures, which are decoded and converted into a wide variety of biochemical changes by Ca2+ sensors. Besides evolutionarily conserved calmodulin (CaM), plants exclusively possess a group of calmodulin-like proteins (CMLs), which play central roles in the coordination of plant responses to different external stimuli. Nevertheless, only few of these proteins have been thoroughly characterized and demonstrated to function as Ca2+ sensors.
Our research is focused on the investigation of the metal-binding, physicochemical and structural properties of various plant CMLs using complementary biophysical and structural approaches in order to correlate their properties with the biological activity.
We have recently characterized CML36 from Arabidopsis thaliana, demonstrating that in vitro the protein shows features consistent with Ca2+ sensor function. ITC analysis revealed that CML36 possesses two high affinity Ca2+/Mg2+ mixed sites and two low affinity Ca2+-specific sites. Binding of Ca2+ to CML36 increases its α-helical content and triggers a conformational change that exposes hydrophobic surfaces necessary for target recognition. Ca2+ and Mg2+ ions also stabilized the tertiary structure of CML36. In particular, cations binding to the Ca2+/Mg2+ mixed sites appears to guide a large structural transition from a loosely packed molten globule apo-state to a well-defined, stable holo-structure. Through in vitro binding experiments, we showed that CML36 directly interacts with the N-terminal domain of Arabidopsis Ca2+-ATPase isoform 8 (ACA8), a type IIB Ca2+ pump localized at the plasma membrane (PM). Moreover, we demonstrated that this interaction promotes ACA8 Ca2+-dependent hydrolytic activity in vitro.
References:
- Astegno A, Maresi E, Bertoldi M, La Verde V, Paiardini A, Dominici P (2017) Unique substrate specificity of ornithine aminotransferase from Toxoplasma gondii. Biochem J. 474(6):939-955.
- Vallone R, La Verde V, D'Onofrio M, Giorgetti A, Dominici P, Astegno A (2016) Metal binding affinity and structural properties of calmodulin-like protein 14 from Arabidopsis thaliana. Protein Sci. 25(8):1461-71.
- Kumar N, Astegno A, Chen J, Giorgetti A, Dominici P (2016) Residues in the distal heme pocket of Arabidopsis non-symbiotic hemoglobins: implication for nitrite reductase activity. Int J Mol Sci. 28;17(5).
- Astegno A, La Verde V, Marino V, Dell'Orco D, Dominici P (2016) Biochemical and biophysical characterization of a plant calmodulin: Role of the N- and C-lobes in calcium binding, conformational change, and target interaction. Biochim Biophys Acta. 1864(3):297-307.
- Astegno A, Capitani G, Dominici P (2015) Functional roles of the hexamer organization of plant glutamate decarboxylase. Biochim Biophys Acta. 1854(9):1229-37.
He Jianwei
Liaoning University, China
Title: Structural insights into the mechanism of how polyphenols suppresses amyloid fibrillation
Biography:
He Jianwei, professor at School of Life Science, Liaoning University, P.R.China, received M.S. degrees in Biochemistry in 2002, from Yamaguchi University, Japan. He completed his Ph.D in Bioresource in 2005 at Tottori University, Japan, his research interests include: 1) Using molecular dynamics and biochemical methods to study protein oligomerization progress and the importance of dimers and tetramers in the aetiology of amyloidotic diseases. 2) Mining, screening or designing of novel inhibitors of natural resources against protein misfolding and amyloid aggregation.
Abstract:
Polyphenols, especially natural flavonols, have received considerable public attention in China due to the positive association between food and traditional herbal consumption and beneficial health effects. Flavonoids has been demonstrated to be active inhibitors of fibrillation by amyloidogenic protein. We recently reported the inhibitory activity of Myricetin against HEWL fibril formation, in which Myricetin exhibited a stronger inhibition than the well-characterized polyphenol Quercetin. In contrast to our previous studies using other polyphenols, we find the generation of irregular structural aggregates formed by the binding of Morin to HEWL, which support a novel and distinctive model for how this small molecule inhibits amyloid formation. Moreover, we also demonstrated that EGCG was a potent inhibitor of amyloidogenic cystatin amyloid fibril formation in vitro. Through combining experimental and computational data, we were able to propose a mechanism by which EGCG inhibited the fibrillation of cystatin: EGCG appears to be a generic inhibitor of amyloid-fibril formation, although the mechanism by which it achieves such inhibition may be specific to the target fibril-forming polypeptide. In conclusion, our findings implicate the importance of diet and drink habits as playing a major role in guarding against amyloid fibril formation and promoting healthy aging.

References:
- Chong Xiaoying, Sun Luchen, Sun Yonghui, Chang Lin, Chang K Alan, Lu Xian, Zhou Xuejie, Liu Junqing, Zhang Bing, Jones W Gary, He Jianwei(*). Insights into the mechanism of how Morin suppresses amyloid fibrillation of hen egg white lysozyme. Int J Biol Macromol. 2017, 101: 321-325.
- Wang Na., He Jianwei(*), Chang Alan K., Wang Yu, Xu Linan, Chong Xiaoying, Lu Xian, Sun Yonghui, Xia Xichun, Li Hui, Zhang Bing, Song Youtao, Kato Akio, Jones Gary W.*. (-)-Epigallocatechin-3-gallate Inhibits Fibrillogenesis of Chicken Cystatin. J Agric Food Chem. 2015, 63(5): 1347-51.
- He Jianwei(*),Wang Yu,Chang Alan K.,Xu Linan,Wang Na,Chong Xiaoying,Li Hui,Zhang Bing,Jones GaryW.(*),Song Youtao(*),Myricetin Prevents Fibrillogenesis of Hen Egg White Lysozyme,J Agric Food Chem. 2014, 62(39): 9442-9449
- Chong Xiaoying, Lu Xian, Wang Yu, Chang Alan K., Xu Linan, Wang Nan, Sun Yonghui, Jones Gary W., Song Youtao, Song Yong-Bo, He Jianwei(*).Distinct structural changes in wild-type and amyloidogenic chicken cystatin caused by disruption of C95-C115 disulfide bond. J Biomol Struct Dyn. 2016, 5:1-9.
- He Jianwei,Xu Linan,Zou Zhiyuan,Ueyama N.,Li Hui,Kato Akio,Jones GW.,Song Youtao(*),Moleculardynamics simulation to investigate the impact of disulfide bond formation on conformational stability ofchicken cystatin I66Q mutant.,J Biomol Struct Dyn.,2013,31(10):1101-111
Federica Cossu
Institute of Biophysics at the National Research Council (IBF-CNR), Italy
Title: Type I BIR domain inhibitors in cancer therapy: designing drugs to modulate the NF-κB pathway
Time :

Biography:
Federica Cossu has always been interested in the field of cancer research, being fascinated by structural studies of crucial macromolecules and protein complexes involved in the cellular processes of cell death/survival. She gathered experience in cloning, expression, purification and crystallization of recombinant proteins, mainly belonging to the field of cancer. During the last years, she focused on the structure-based design of small molecules to be developed as drug candidates directed to pre-clinical studies. The success of this activity is proven by one patent, one award for her PhD thesis and several publications in the field. She progressively improved her knowledge on biophysical techniques for the study of proteins and on the in silico analysis of protein structures. She collected experiences in European laboratories, including several short visits/experiments at the ESRF synchrotron in Grenoble and at SOLEIL in Paris. She has been working in lab equipe for ten years, covering different positions within the research group, from master degree student to post-doc. She has been the supervisor of students, also giving lessons on the structural approaches applied to cancer therapy.
Abstract:
Inhibitors of apoptosis proteins (IAPs) constitute a family of conserved proteins whose over-expression enhances cell survival and resistance to anticancer agents. IAPs are E3 ligases, ubiquitylating substrates for the regulation of NF-kB; furthermore, they sequester caspases to prevent apoptosis. IAPs interactions occur through type I and type II BIR (Baculovirus IAP repeat) domains. Smac-mimetics (SM) mimicking the active N-terminal peptide of Smac-DIABLO, the natural antagonist of IAPs, have been shown to sensitize cancer cells to apoptosis. SM interact with type II BIR domains of IAPs, thus relieving caspases from X-linked IAP (XIAP) inhibitory activity and leading to cellular IAPs (cIAPs) auto-ubiquitylation and proteasomal degradation within minutes from exposure. Although SM are currently promising candidates for cancer therapy, some cancer cell lines present SM-resistance due to renewed cIAP2 activity and re-activation of NF-κB. IAPs-mediated regulation of NF-kB signaling is based on the formation of different protein-protein complexes, regulating ubiquitin-dependent signal transduction cascades. The type I BIR domain from different IAPs has been recognized as a pivotal platform for the assembly of such complexes.
We analyzed the surface of type I BIR domains (X- and cIAP-BIR1) to identify the hot-spots for the relevant protein-protein interactions. Virtual docking using libraries of compounds returned hits (NF023 and analogues) able to impair BIR1-based complexes with predicted low micromolar affinities that were experimentally confirmed. To this purpose, in vitro assays include fluorescence-based and biophysical techniques (Thermofluor, Microscale Thermophoresis, SEC, DLS, SLS). Crystallography on the protein-ligand complexes is the core of the structure-driven approach used for the iterative optimization of specific and selective drug candidates. Treatment of cancer cell cultures with the selected compounds will verify their effects on the modulation of IAPs-dependent signaling cascades. This represents a novel strategy to promote apoptosis in cancer and will unravel new insights on the regulation of NF-kB pathway.
References:
- Federica Cossu, Milani M, Grassi S, Malvezzi F, Corti A, Bolognesi M and Mastrangelo E (2015) “NF023 binding to XIAP-BIR1: Searching drugs for regulation of the NF-κB pathway”. Proteins 83(4): 612-20
- Workshop by NanoTemper Technologies GmbH
Session Introduction
Tobias Pflüger
NanoTemper Technologies GmbH, Germany
Title: Solutions for studying protein complexes in structural biology and drug development
Biography:
Tobias Pflüger studied Chemistry at the Albert-Ludwigs-University in Freiburg with an emphasis on biochemistry. He received his PhD in Structural Biology investigating the structure and function of membrane proteins involved in cellular signalling cascades. In late 2015, Tobias joined NanoTemper Technologies as an application specialist.
Abstract:
Key to understanding the functional role of target proteins is elucidating the structure-function relationship between two proteins or protein complexes. Current analysis is often complex, time-consuming, and lacks data quality.
One of the goals of structural biology is to bring together multiple disciplines and methodologies as a means to better characterize proteins and protein complexes. For highly utilized techniques such as cryo-EM, X-ray crystallography and NMR, having the ability to identify and quantify binding affinities as well as analyze conformational and colloidal properties, enables researchers to gain a better understanding of the functional properties of protein targets.
During this workshop, you will learn about practical solutions to monitor and optimize protein stability and quality. An overview of the various analytical and biophysical methods commonly used by structural biologist will be discussed. We will share case studies demonstrating the benefits of understanding conformational and colloidal properties of protein complexes and how to use this information for further downstream analysis. Finally, we will share examples of protein-small molecule and protein-protein interactions and novel methodologies that assist researchers in making better decisions.