

Christina Scharnagl
Technical University of Munich, Germany
Title: Does the dynamics of their transmembrane domain qualify bitopic membrane proteins as substrates for intramembrane proteolysis?
Biography
Biography: Christina Scharnagl
Abstract
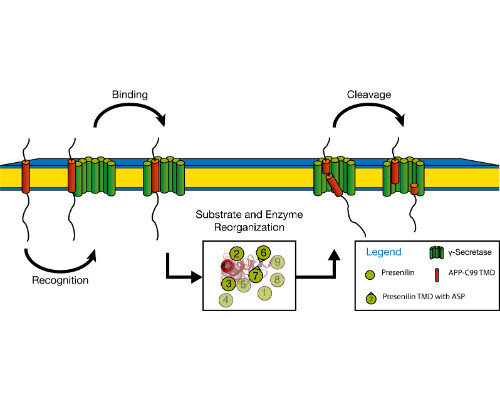
Integral membrane proteins facilitate communication between the inside of the cell and its exterior. Their transmembrane domains (TMDs) support a diversity of biological functions and exhibit sequence-dependent conformational dynamics on multiple size and time scales. Membrane proteins are notoriously difficult to study by experimental methods. Molecular dynamics (MD) simulations provide a powerful tool of high spatial and temporal resolution that effectively complements experimental methods. Here we focus on the conformational dynamics of the TMD of the amyloid precursor protein (APP). APP is enzymatically hydrolyzed within its TMD by g-secretase (GSEC), forming toxic Ab peptides regarded as molecular cause of Alzheimer's disease (AD). Besides APP, GSEC cleaves ~100 single-span membrane proteins within their TMDs, however without obvious consensus sequence. Finding the link between the molecular architecture of the substrate TMDs and cleavage is, therefore, of upmost importance. Because unfolding is obvious to expose the scissile bond, it seems plausible that the TMD itself is optimized for local helix unwinding. However, this view was challenged by our experiments and MD simulations. Our results suggest an alternative model where reaching a cleavage-competent state involves multiple conformational transitions of the substrate/enzyme complex where global conformational plasticity of the substrate TMD is a key determinant. In a first step, we compare the conformational flexibility of a large number of substrate and non-substrate TMDs, as well as TMDs carrying missense mutations related to early onset AD. Knowing the key-dynamical motifs will help to identify new substrates and to elucidate the physiological functions of the protease in the brain and other organs. This work is part of a collaborative research program (https://www.i-proteolysis.de/).
References:
- Langosch C, Scharnagl C, Steiner H, Lemberg M (2015) Understanding intramembrane proteolysis: from protein dynamics to reaction kinetics. Trends Biochem. Sci. 40:318-327.
- Scharnagl C, Pester O, Hornburg P, Hornburg D, Götz A, Langosch D (2014) Side-chain to main-chain hydrogen bonding controls intrinsic backbone dynamics of the amyloid precursor protein transmembrane helix. Biophys. J. 106: 1318-1329.
- Pester O, Götz A, Multhaup G, Scharnagl C, Langosch D (2013) The cleavage domain of the amyloid precursor protein transmembrane helix does not exhibit above-average backbone dynamics. ChemBioChem 14:1943-1948.
- Pester O, Barrett P, Hornburg D, Hornburg P, Pröbstle R, Widmaier S, Kutzner C, Dürrbaum M, Kapurniotu A, Sanders CR, Scharnagl C, Langosch D (2013) The backbone dynamics of the amyloid precursor protein transmembrane helix provides a rationale for the sequential cleavage mechanism of g-secretase. J. Am. Chem. Soc. 135: 1317-1329.
- Ried C, Scharnagl C, Langosch D (2015) Entrapment of water at the transmembrane helix-helix interface of quiescin sulfhydryl oxidase 2. Biochemistry 55: 1287-1290.